


Анализ последовательностей ДНК проливает свет на биологию микроорганизмов, но оценить функции генов, которые имеют мало или вообще не имеют сходства с охарактеризованными генами, довольно сложно. Ученые обсуждают эту проблему с геномной и микробной точек зрения.
Структурирование и контекст в генетических задачах
Функция большинства микробных генов неизвестна. Некоторые гены из этой микробной "темной материи" могут кодировать ранее неизвестные типы ферментов или классы антибиотиков. По мере того как все больше генов с неизвестной функцией обнаруживается с помощью секвенирования ДНК из совокупностей нескольких геномов, называемого метагеномным секвенированием, трудности с экспериментальной характеристикой этих загадочных генов привели к тому, что основное внимание стало уделяться вычислительному прогнозированию их функций.
Две публикации в журнале Nature - одна Rodríguez del Río et al., а другая Pavlopoulos et al., опубликованная в октябре прошлого года, - решают эту задачу, умело используя достижения алгоритмов кластеризации (вычислительных инструментов, которые группируют гены на основе сходства аминокислотных последовательностей) и инструментов предсказания структуры белка, таких как AlphaFold.
Несмотря на различия в технических подходах, основная стратегия, использованная Павлопулосом и Родригесом дель Рио, была схожей. Они кластеризовали сотни миллионов белковых последовательностей из массивов метагеномных данных в ранее неизвестные белковые семейства. Однако Родригес дель Рио и его коллеги отфильтровали свои данные, чтобы изучить гены только прокариот (организмов, клетки которых не имеют ядра), в то время как Павлопулос и др. использовали данные, которые также включали последовательности эукариот (организмов, клетки которых имеют ядро) и вирусов.
Имея под рукой каталоги ранее неизвестных семейств, обе группы задались целью предсказать функции своих новых описанных семейств, используя геномно-контекстный анализ, который предполагает изучение соседних генов для получения информации о функциях, а также используя достижения в методах предсказания структуры белков. В прокариотических геномах гены, участвующие в одном и том же процессе, часто располагаются близко друг к другу. Геномно-контекстный анализ, предполагающий "соседство по ассоциации", был эффективно использован для предсказания ранее неизвестных систем противовирусной защиты, используемых бактериями. Второй подход - сравнение предсказанных структур белков для поиска похожих (гомологичных) белков - более чувствителен, чем простое сравнение аминокислотных последовательностей. Обе группы ученых предсказали структуры для своих семейств белков и сравнили их с базами данных известных структур, тем самым сделав обоснованные прогнозы относительно функций некоторых из этих загадочных белков.
Масштабы и вычислительные затраты, вложенные в эти работы, в результате которых были обнаружены сотни тысяч новых белковых семейств (иллюстрация 1), впечатляют. Тем не менее, количество ранее неизвестных генов, имеющих функциональное предсказание, по-прежнему остается относительно небольшим. В обеих публикациях только около 15% ранее неизвестных семейств белков могли быть аннотированы на основе структурного сходства; анализ геномного контекста позволил предложить функции для 7,4% семейств в работе Павлопулоса и 13% в работе Родригеса дель Рио. Кроме того, некоторые обозначенные функциональные категории (например, "рибосома") не получили детального описания, что может затушевывать точную роль этих генов. В конечном счете, надежность этих предсказаний должна быть определена экспериментально. В действительности, Родригес дель Рио и др. сделали первый шаг к этой цели, экспериментально проверив аннотацию для двух из предсказанных ими семейств.
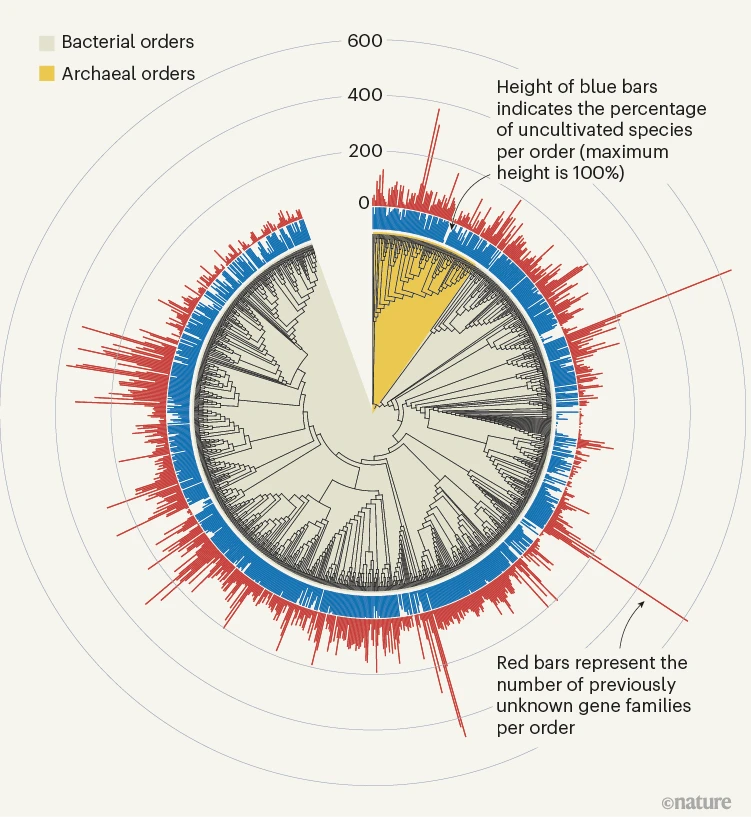
Иллюстрация 1. Ранее неизвестные семейства микробных генов. Крупномасштабный анализ последовательностей ДНК, полученных из образцов микроорганизмов, о котором сообщали Родригес дель Рио и Павлопулос, позволил выявить сотни тысяч ранее неизвестных семейств генов. Эти данные, полученные от микробов, обитающих в дикой природе и различных средах обитания, и включающие виды, которые не культивировались в лабораторных условиях, являются отправной точкой для понимания неизученных аспектов биологии бактериальных и архейных микроорганизмов.
Углубляясь в темную материю микроорганизмов, эти два исследования открывают богатство ранее скрытых знаний, прокладывая путь к будущим открытиям в различных областях - от медицины до биотехнологий. Последующие эксперименты могут включать изучение семейств белков с совершенно новыми белковыми конфигурациями, что, возможно, откроет неизученные биологические функции. Аналогичным образом, синапоморфные гены - соответствующие семействам белков, которые характерны для группы организмов, имеющих общего предка, но отсутствуют у других, - могут дать ключ к разгадке ключевых эволюционных процессов. При дальнейшем совершенствовании и проверке эти вычислительные подходы предлагают мощный инструмент для раскрытия функциональных секретов невидимого микробного мира.
Микробные последовательности раскрывают экологию и эволюцию
Гены - это основной источник всей биологической информации на Земле, от цвета глаз человека до формы клеток микроорганизмов. Кодируемые ими белки можно сгруппировать с помощью биоинформатики в семейства, обычно имеющие общую функциональность. Совокупность всех известных белков в базах данных постоянно расширяется по мере секвенирования геномов и предсказания функций кодируемых ими белков. Наибольшая доля биологического функционального разнообразия на нашей планете приходится на микробные белки. С появлением секвенирования смешанных микробных геномов из окружающей среды (такой подход, позволяющий исследовать несколько геномов, называется метагеномикой) скорость добавления данных в базы данных геномов и белков просто поражает. Однако функциональные возможности большинства семейств белков неизвестны и являются частью микробной темной материи.
В работе Родригеса дель Рио и коллег, а также в исследовании Павлопулоса и др. проанализированы масштабные метагеномные данные и изучены потенциальные функции и распространение неизвестных семейств белков, которые могут иметь эволюционное и экологическое значение. Родригес дель Рио проанализировал около 150 000 микробных геномов, а Павлопулос с коллегами исследовали почти 27 000 совокупностей метагеномных данных, полученных из различных экосистем с помощью различных биоинформационных подходов, что значительно превышает масштаб записей в публичных базах данных, использовавшихся в предыдущих подобных исследованиях. Удивительно, но метод, названный Павлопулосом и его коллегами анализом разреженности, не выявил замедления в обнаружении ранее неизвестных семейств белков по мере добавления новых метагеномов в анализ. Напротив, количество обнаруженных семейств белков росло в геометрической прогрессии, что послужило основанием для проведения целого ряда последующих исследований.
Распределение семейств белков по категориям экосистем Земли (биомам), представленное Павлопулосом и коллегами, подтверждает результаты предыдущих исследований, касающихся распределения микробных генов. Однако некоторые биологические объекты оказались особенно богатыми источниками вновь открытых семейств белков, включая вирусы, о которых сообщают Павлопулос и др. Последние представляют собой группу микроорганизмов, называемых археями, которые находятся в близком родстве с первым предком эукариот. Таким образом, изучение их белков может открыть новые горизонты в эволюции эукариотической клетки.
Одной из основных проблем при изучении множества ранее неизвестных семейств белков, закодированных в геномах природных образцов, является идентификация эукариотических генов в метагеномах. Хотя существуют определенные алгоритмы для извлечения эукариотических геномов из метагеномов, точное предсказание эукариотических генов в смешанных последовательностях ДНК - эквивалент метода Павлопулоса и коллег по идентификации микробных генов - все еще невозможно биоинформационно. Как только этот недостаток будет преодолен с помощью новых алгоритмов, ученые существенно расширят "пространство последовательностей" белков и выявят семейства белков с неизвестными функциями, которые определяют экологию и эволюцию эукариот.
Наибольший прогресс в кропотливой организации белковых семейств почти 27 000 метагеномов и всего древа жизни заключается в выявлении экосистемно-специфических белковых кластеров, которые отличаются по своему присутствию или отсутствию, или относительному обилию между различными условиями данной экосистемы - например, между контекстами здоровья или заболевания. Применив эту стратегию для изучения микробных данных здоровых людей и больных колоректальным раком, Родригес дель Рио и его коллеги обнаружили, что в бактериях кишечника людей, больных раком, были обогащены определенные неизвестные семейства белков. Эти семейства белков были связаны с микробной подвижностью, адгезией и инвазией в тканях человека, что было выявлено с помощью геномно-контекстного анализа. Использование этого подхода в других областях исследований должно быть чрезвычайно полезным для расшифровки различных функций образцов в надежде выявить новые мишени для биохимических анализов, чтобы с их помощью изучить крошечную часть темной материи микроорганизмов.
Выявление различий в микробных сообществах (микробиомах), которые могут объяснить, например, состояние заболевания человека, в значительной степени зависит от сравнения присутствующих видов и их обилия (таксономический состав), а также от изучения генов, связанных с определенными функциями. Поиск специфических, но дифференциально распространенных семейств белков с неизвестной функцией, как продемонстрировали Родригес дель Рио и соавторы, может не только заменить существующие подходы к дифференциации микробиомов на основе маркерных генов, но и вывести исследования микробиома на новый, обусловленный причинно-следственными связями уровень.









