

Под микроскопом ткани млекопитающих демонстрируют свою сложную и элегантную архитектуру.
Но если вы посмотрите на ту же ткань после образования опухоли, вы увидите бедлам. Итай Янаи, вычислительный биолог из Нью-Йоркского университета, пытается найти порядок в этом хаосе. "Существует особая логика в том, как все устроено, и пространственная транскриптомика помогает нам увидеть это", - говорит он.
Пространственная транскриптомика - это общий термин, охватывающий более десятка методов для составления схемы экспрессии генов на уровне генома в образцах тканей, разработанных в дополнение к методам секвенирования РНК в единичных клетках.
Однако у этих методов секвенирования единичных клеток есть недостаток - они могут быстро профилировать содержание РНК мессенджера (или транскриптома) большого количества отдельных клеток, но обычно требуют физического нарушения исходной ткани, что приводит к потере важной информации о том, как организованы клетки, и может изменить их таким образом, что это может запутать последующие исследования. Иммунолог Идо Амит из Научного института Вейцмана, Израиль, говорит, что такие эксперименты иногда заставляют его группу сомневаться в своих результатах. "Действительно ли это состояние in situ, или мы просто смотрим на что-то, что либо не является основным [фактором], либо вообще не существует?".
В отличие от этого, пространственная транскриптомика позволяет исследователям изучать экспрессию генов в неповрежденных образцах, открывая новые горизонты в исследовании рака и выявляя ранее недоступную биологию хорошо изученных тканей. Полученные "атласы" пространственной информации могут рассказать ученым о том, какие клетки составляют каждую ткань, как они организованы и как они общаются. Но составить такие атласы нелегко, поскольку методы пространственной транскриптомики обычно представляют собой противоречие между двумя конкурирующими целями: более широким охватом транскриптома и более высоким пространственным разрешением. Развитие экспериментальных и вычислительных методов в настоящее время помогает исследователям сбалансировать эти цели - и улучшить клеточное разрешение в процессе.
Масштабирование FISH
Корни пространственной транскриптомики восходят к 1960-м годам и развитию гибридизации in situ. Этот метод использует меченые фрагменты нуклеиновой кислоты в качестве зондов для определения наличия и расположения комплементарных последовательностей ДНК или РНК в клетках или тканях. Первоначально исследователи использовали радиоактивные метки, но позже перешли на флуоресцентные метки, которые можно визуализировать под микроскопом.
К 1998 году, благодаря достижениям в области микроскопии и обработки изображений, исследователи смогли идентифицировать отдельные молекулы РНК в клетках. Используя метод одномолекулярной флуоресцентной гибридизации in situ (smFISH), можно было визуализировать отдельные транскрипты мРНК нескольких генов одновременно с помощью зондов разного цвета. Но ранние версии smFISH могли одновременно отслеживать только три или четыре гена, что далеко не соответствовало десяткам тысяч генов, экспрессируемых в транскриптоме человека. "Одно из фундаментальных ограничений микроскопии заключается в том, что вы не можете одновременно наблюдать за таким количеством цветов или молекул, хотя и получаете очень богатую пространственную информацию", - говорит Фей Чен, клеточный биолог из Массачусетского технологического института.
Умные усовершенствования техники с тех пор позволили преодолеть эти ограничения. Например, мультиплексный FISH (MERFISH), о котором в 2015 году сообщили биофизик Сяовэй Жуань и ее коллеги из Гарвардского университета, позволяет обнаруживать и различать тысячи транскриптов мРНК различных генов с помощью всего нескольких флуоресцентных меток. Каждому транскрипту присваивается уникальный двоичный штрих-код, состоящий из единиц и нулей, а затем он маркируется несколькими комплементарными "кодирующими зондами", содержащими считываемые сиквенсы. Затем образцы проходят последовательные циклы гибридизации и визуализации с различными флуоресцентно меченными "считывающими зондами" для расшифровки этого штрих-кода.
Когда считывающий зонд связывается со считываемым сиквенсом кодирующего зонда и дает флуоресцентный сигнал, он считывается как "1"; если флуоресценция отсутствует, он считывается как "0". В результате нескольких раундов визуализации получается двоичный штрих-код, который позволяет идентифицировать обнаруженную РНК. Метод "защиты от ошибок" относится к конструкции штрих-кодов: они достаточно сильно отличаются друг от друга, поэтому вероятность неправильной интерпретации последовательности мРНК очень мала.
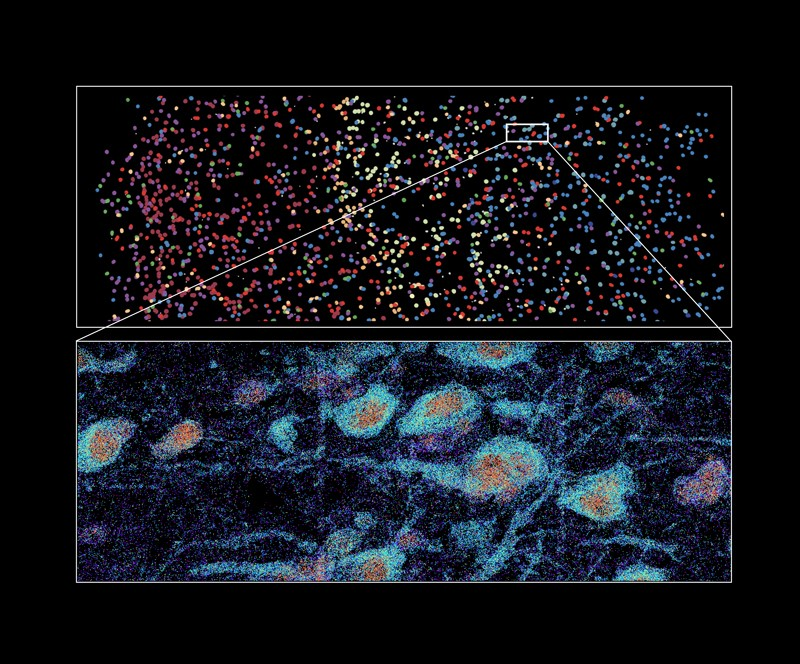
Изображение MERFISH части человеческого мозга, показывающее типы клеток, помеченные различными цветами (вверху) и помеченные молекулы РНК различных генов в отдельных клетках (внизу).Фото: Xiaowei Zhuang Lab, Harvard University and HHMI.
Хотя изначально метод был описан как инструмент для одноклеточного анализа, команда Жуань также применяет его к тканям, включая человеческий мозг. "Профилируя экспрессию 4 000 генов, мы смогли создать молекулярно определяемый клеточный атлас коры головного мозга человека с беспрецедентным молекулярным и пространственным разрешением", - говорит она. Этот анализ, результаты которого опубликованы в журнале Science, позволил установить идентичность и расположение более 100 различных подтипов клеток, а также выявить поразительные различия в клеточном составе и организации структур коры головного мозга человека". В более ранней работе группа Жуань также использовала этот метод для картирования частей мозга мыши, включая моторную кору и гипоталамус.
Другие методы баркодирования и визуализации дают аналогичные преимущества. Например, картирование ампликонов транскриптов с пространственным разрешением (STARmap), описанное группой специалистов Стэнфордского университета в 2018 году, использует форму секвенирования in situ для обнаружения транскриптов мРНК в интактных образцах тканей. Используя набор генно-специфических штрих-кодов, каждый из которых состоит из 5 нуклеотидов, специалисты Стэнфорда отобразили и количественно определили более 1000 транскриптов генов в тканях мозга мыши с разрешением в одну клетку.
Однако у методов, основанных на визуализации, есть и недостатки. Например, по мере того, как эти методы охватывают все больше целей, они становятся все более трудоемкими. MERFISH может одновременно определять более 10 000 генов, но эксперименты такого масштаба обычно требуют дополнительного этапа - "протокола экспансии ткани" для увеличения объема каждого образца, чтобы микроскопия могла успешно различать различные молекулы. Другой метод, seqFISH+, преодолевает это ограничение за счет использования более сложной стратегии цветового кодирования. Но seqFISH+ требует гораздо больше раундов маркировки и визуализации - 80, по сравнению с 23 для MERFISH - для того же количества генов. И оба метода требуют более одного дня непрерывной микроскопии для сбора данных в масштабе транскриптома.
Массив альтернатив
Возможно, самым главным ограничением методов, основанных на гибридизации, является то, что исследователи должны заранее решить, на какие гены они хотят нацелиться. "Как только вы начинаете выбирать маркеры, вы теряете информацию", - говорит Амит. Методы на основе массивов предлагают более широкий взгляд на транскриптомы, но за это приходится платить - они имеют более низкую чувствительность и меньшее пространственное разрешение.
Йоаким Лундеберг, молекулярный генетик из Королевского технологического института в Стокгольме, который является одним из пионеров пространственной транскриптомики, описал такой подход в 2016 году. Он и его коллеги покрыли стеклянное предметное стекло упорядоченным массивом олигонуклеотидов, предназначенных для захвата нитей мРНК. Они работают путем связывания с длинным хвостом адениновых нуклеотидов, которым заканчивается каждый транскрипт мРНК. После нанесения тонкого среза ткани на верхнюю часть предметного стекла исследователи обработали ткань химическими веществами, которые сделали ее проницаемой, позволив РНК просочиться наружу и связаться с массивом. Затем захваченная РНК была преобразована в ДНК и секвенирована. Поскольку каждый олигонуклеотид содержит характерный штрих-код, обозначающий его положение на предметном стекле, итоговые данные показывают не только идентичность мРНК, но и ее расположение в ткани. Полученные данные можно представить в виде пиксельной карты, наложенной на микроскопическое изображение, где каждый пиксель показывает, какие гены были экспрессированы в каждой позиции.
Команда Лундеберга использовала этот метод для анализа полного транскриптома образцов тканей мозга и опухолей, хотя и с ограниченным пространственным разрешением. В оригинальном методе пиксели описывали участки диаметром около 100 микрометров - в 10 раз шире, чем типичная клетка. С тех пор этот метод был коммерциализирован фирмой 10x Genomics как платформа Visium Spatial Gene Expression с размером участка 55 мкм. Группа Янаи использовала эту платформу для картирования архитектуры опухолей поджелудочной железы и кожи. И даже без разрешения в одну клетку они получили ценные сведения об архитектуре опухоли и биологически важных взаимодействиях между раковыми клетками, здоровыми тканями организма и популяциями иммунных клеток, рассказывает он.
В последние несколько лет наблюдается шквал работ по повышению разрешающей способности методов на основе массивов. Чен и его коллега Эван Макоско, например, разработали метод под названием Slide-seq, разрешение которого составляет 10 мкм - примерно размер одной клетки, говорит Чен. Компания 10x Genomics объявила, что ее платформа нового поколения Visium HD, которая должна быть выпущена в конце этого года, также будет обеспечивать разрешение в одну клетку, хотя никаких данных пока не опубликовано.
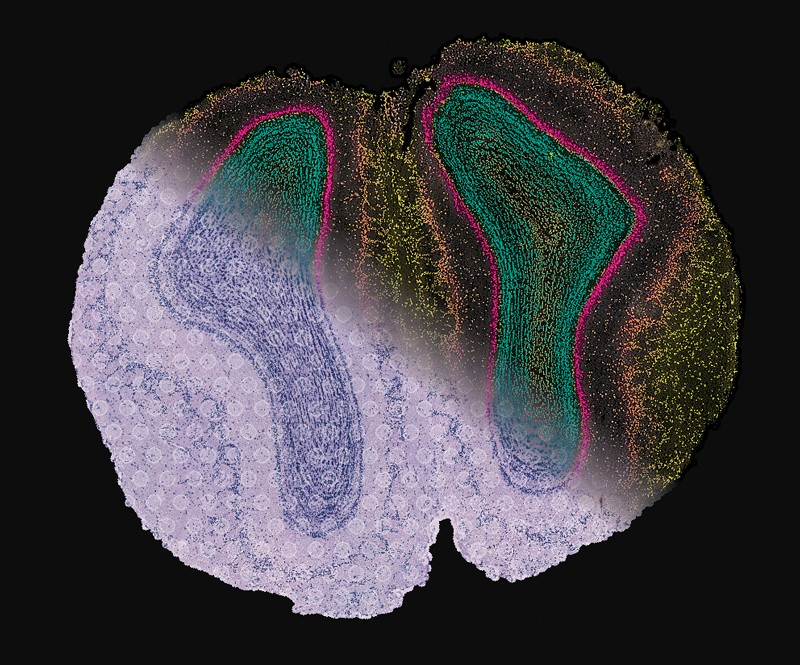
Объединенное изображение, показывающее структуру ткани, данные РНК и карты экспрессии генов с клеток в обонятельной луковице мыши со сверхразрешением. Фото: Ludvig Bergenstråhle
В мае исследователи из компании BGI, Китай, описали метод, основанный на массивах, который позволяет преодолеть барьер одной клетки. Он называется Stereo-seq и использует узорчатые массивы наношариков ДНК со штрих-кодом, диаметр которых составляет примерно 200 нанометров, а расстояние между ними - несколько сотен нанометров. "Для генерации данных по одной клетке у нас фактически есть что-то около 400 точек данных", - говорит Сюнь Сюй, исполнительный директор BGI Group и один из разработчиков метода. Его можно применять к большим образцам, включая целый мозг макаки, который был разрезан на кусочки размером три на пять сантиметров, о чем сообщается в препринте этого года. Только на секвенирование ушло почти два месяца, говорит Ао Чен из BGI, который также является членом команды Stereo-seq.
Но с повышением разрешения растут и технические проблемы. Одна из них - диффузия: когда мРНК вытекают из ткани, они могут распространяться в стороны, прежде чем встретить зонд захвата, что искажает данные. Лундеберг говорит, что, оптимизируя степень пермеабилизации ткани, исследователи могут ограничить эту диффузию до нескольких микрометров, что более чем достаточно для клеточного разрешения. "Если вы действительно хотите увидеть субклеточное разрешение, вам следует обратиться к платформам на основе визуализации", - предлагает он.
Другая проблема связана с физикой: с уменьшением размера пикселя уменьшается и количество зондов, доступных для захвата мРНК. Лундеберг говорит, что он отказался от версии платформы своей группы с высоким разрешением, потому что ей не хватало чувствительности для захвата биологически значимых сигналов мРНК. BGI сообщает, что Stereo-seq обычно позволяет обнаружить 300-500 генов на клетку, что дает полезное, но ограниченное представление об активности экспрессии генов. Несмотря на это, команда использовала метод для построения 3D атласов, которые показывают пространственные сдвиги в экспрессии генов, сопровождающие эмбриональное развитие у мышей, мух и рыбок зебрафиш.
Чтение между строк
Для осмысления пространственных данных требуются специальные вычислительные инструменты. Например, исследователям может понадобиться определить, какие типы клеток присутствуют в организме, используя данные, содержащие только часть транскриптома. Многие исследователи добиваются этого путем параллельного анализа данных секвенирования РНК одной клетки, собранных из одной и той же ткани. "Затем вы можете сопоставить и согласовать то, что вы видите в пространственных данных, с тем, что вы видите в одноклеточных данных", - говорит Фей Чен. Такое сопоставление позволяет исследователям наносить на пространственные транскриптомные карты типы клеток, выведенные из совокупности данных РНК-секвенирования.
Некоторые алгоритмы могут даже определять клеточный состав относительно крупных пикселей, получаемых такими платформами, как Visium, которые могут содержать множество клеток. Фей Чен и специалист по вычислительной биологии из Гарварда Рафаэль Иризарри разработали алгоритм с открытым исходным кодом под названием "надежное разложение по клеточному типу" (RCTD). RCTD, называемый также клеточной деконволюцией, широко применим к большинству методов, основанных на массивах, говорит Фей Чен. Он не только определяет, какие клетки присутствуют в данном пикселе, но и дополняет недостающие детали об активности экспрессии генов этих клеток. RCTD может применяться к методам визуализации, таким как MERFISH, для сегментации, добавляет Фей Чен, - определения клеточных границ по данным экспрессии генов, полученным в результате секвенирования РНК одной клетки.
Данные визуализации также могут стать мощным подспорьем для клеточной деконволюции, а большинство методов пространственной транскриптомики на основе массивов могут собирать такие данные параллельно, считает Мингяо Ли, генетик и статистик из Университета Пенсильвании. "Вы можете увеличить масштаб, посмотреть на тканевые особенности, сколько клеток, какова плотность клеток и каковы морфологические особенности отдельных клеток", - говорит она. Но связать эти элементы воедино - сложная задача, требующая большого объема данных и зачастую сложных вычислительных подходов.
Например, Лундеберг с коллегами опубликовали исследование , в котором они обучили алгоритм глубинного обучения транскриптомным и гистологическим данным с системы Visium, чтобы экстраполировать детали, выходящие далеко за пределы содержимого отдельных участков. "Мы могли очень точно предсказывать экспрессию генов между участками", - говорит он, имея в виду физические разрывы, которые присущи каждому методу, основанному на массивах. "Из этого можно сделать вывод о разрешающей способности одной клетки".
Однако определение типов клеток - это только начало. Различные типы клеток могут иметь поразительно разные фенотипы в зависимости от того, где они расположены в ткани, и эти паттерны дифференциальной экспрессии генов могут сделать пространственный клеточный атлас гораздо более мощным. Алгоритмы машинного обучения также полезны для выявления этой изменчивости. Например, Амит и его коллеги разработали метод под названием DestVI, который позволяет определить, какие клетки расположены в каждой точке массива, и уловить отличительные биологические состояния различных типов клеток. Используя его, исследователи определили фенотипы иммунных клеток в раковых тканях. "Можно достичь гораздо более высокого уровня понимания физиологии или патологии в ткани", - говорит Амит.
Свести все воедино
Возможно удивительно для области, которая производит так много данных, но исследователям пространственной транскриптомики сейчас нужно еще больше данных. Особенно ценны такие инициативы, как Атлас клеток человека, в рамках которого были опубликованы транскриптомные данные, собранные из миллионов клеток 33 органов (www.humancellatlas.org). Такие высококачественные, стандартизированные данные могут быть использованы, например, для обучения аналитических алгоритмов.
Пространственная транскриптомика еще не достигла того уровня сотрудничества и обмена данными, который наблюдается в более развитых областях, таких как геномика или транскриптомика единичных клеток, и это может стать источником разочарования. Во многих случаях, говорит Фей Чен, лаборатории делятся только минимумом - необработанными данными эксперимента, что означает, что на воспроизведение работы могут уйти месяцы. Но есть и многообещающие достижения. Например, после публикации результатов работы по стерео-секвенированию BGI создала Консорциум пространственно-временной омики, в котором уже участвуют более 80 исследователей со всего мира. Его цель - использовать различные пространственные методы для решения сложных вопросов в областях, связанных с физиологией человека, патогенезом и эволюционной биологией.
В то же время исследователи стремятся к дальнейшему совершенствованию технологии. Например, команда Лундеберга использует пространственную транскриптомику, чтобы сделать вывод о геномных изменениях, происходящих во время развития опухоли предстательной железы, которые обычно доступны только при секвенировании генома изолированных клеток. "В пределах одного участка ткани вы видите эти чрезвычайно ранние события, которые никто не замечает", - говорит он, добавляя, что многие из этих изменений происходят в клетках, которые в остальном кажутся доброкачественными.
Что касается Янаи, то он в восторге от возможности подслушать, как соседние клетки общаются друг с другом и влияют друг на друга. Такое взаимодействие является важным компонентом нормального формирования и развития органов и может помочь раскрыть принципы организации опухолевой ткани. "Раковые клетки манипулируют нераковыми клетками, - говорит Янаи. Пространственная транскриптомика может зафиксировать эти манипуляции в процессе их осуществления". "Это как недостающий фрагмент головоломки", - говорит он.









