

Сепсис - это синдром, вызванный иммунной дисрегуляцией ответа на инфекцию и являющийся ключевым фактором глобальной смертности от заболеваний.
1. Введение
Сепсис является критическим нарушением функции органов, вызванное дисрегуляторным ответом на инфекцию, что представляет собой серьезную проблему не только для клиницистов, но и для научных исследователей. Воспалительная реакция, повреждение тканей, отказ важных органов и патологическое тромбообразование - основные и типичные патофизиологические изменения при сепсисе, свидетельствующие о наличии инфекции в организме.
При сепсисе происходит выброс многочисленных цитокинов, включая TNF-α, интерлейкины, простагландины и так далее. В то же время лабораторные анализы показывают увеличение количества лейкоцитов, C-реактивного белка и прокальцитонина, повышенный уровень лактата и нарушение функции свертывания кров. Патогенез сепсиса остается недостаточно изученным, что потенциально объясняется преувеличенной воспалительной реакцией и подавлением иммунитета в результате дисрегуляции реакции на инфекцию. Наиболее распространенными симптомами являются лихорадка, учащенное сердцебиение и гипотония, которые могут прогрессировать до возникновения септического шока, отказа многих органов и ДВС (диссеминированного внутрисосудистого свёртывания) -синдрома, представляющих угрозу для жизни.
Стоит отметить, что своевременное распознавание и лечение сепсиса имеют решающее значение для прогноза пациента. Даже при отсутствии четкого диагноза начало противоинфекционной терапии может снизить вероятность возникновения сепсиса. Из-за различных причин и симптомов сепсиса каждый пациент с сепсисом должен проходить индивидуальное лечение в соответствии с его потребностями, клиническими характеристиками и другими параметрами плана лечения. Цель лечения - повысить качество жизни пациента и предотвратить возможные осложнения. Многие стратегии лечения, включая антибиотики, вазоактивные препараты, глюкокортикоиды и иммуномодулирующие средства, являются препаратами первой линии при сепсисе, но часто их необходимо сочетать с другими стратегиями лечения для дальнейшего улучшения клинических симптомов.
В обзоре обобщены потенциальные молекулярные механизмы, лежащие в основе патофизиологии сепсиса и взаимодействия между различными клетками. Кроме того, освещаются последние достижения в области потенциальных биомаркеров и терапевтических препаратов для лечения сепсиса, чтобы предоставить клиницистам и исследователям лучшие идеи и текущий прогресс в исследованиях.
2.Патогенез сепсиса
Основная этиология сепсиса включает бактериальные, грибковые и вирусные инфекции. Бактерии являются наиболее распространенной причиной сепсиса и основными бактериями, вызывающими сепсис, являются золотистый стафилококк (грамположительный) и кишечная палочка (грамотрицательная). Вирусный сепсис встречается реже, чем бактериальный, однако высокая частота встречаемости вирус-отрицательного сепсиса предполагает, что может быть много недиагностированных случаев вирусного сепсиса.
Наиболее распространенными вирусными патогенами в тропических регионах являются вирусы гриппа и денге, оба из которых могут вызывать сезонные вспышки, причем в группе риска находятся новорожденные, дети, беременные женщины, пожилые люди и пациенты с ослабленным иммунитетом. Частота встречаемости грибкового сепсиса выше, чем вирусного, но значительно ниже, чем бактериального. Основным грибковым патогеном при сепсисе является Candida albicans, которая ассоциируется с относительно высоким уровнем смертности. Хотя паразитарные инфекции (<1 %) могут приводить к сепсису, предполагаемая распространенность этого этиологического подкласса остается крайне редкой.
Микробиологию пациентов с сепсисом можно также классифицировать по источнику инфекции, включая внебольничную и внутрибольничную (пациенты, которые не имели инфекции при поступлении, но у них развилась инфекция через 48 ч или более после поступления), причем внутрибольничный сепсис является более тяжелым и связан с более высоким уровнем смертности.
Сепсис характеризуется двумя основными фазами: гипериммунной и иммуносупрессивной, инициируемыми действиями различных иммунных клеток, запускающих серию иммунных ответов. В начальной фазе гиперинтенсивного воспаления повышение уровня провоспалительных цитокинов, выделяемых воспалительными клетками, наряду с активацией систем комплемента и коагуляции, приводит к чрезмерному воспалению, кульминацией которого является цитокиновый шторм и синдром множественной органной дисфункции (MODS). В этот момент происходит преобладание нейтрофилов, активация лимфоцитов, макрофагов и дендритных клеток.
Одновременно или впоследствии увеличивается высвобождение противовоспалительных цитокинов и коингибиторных молекул, снижается экспрессия HLA-DR, происходит гибель иммунных клеток и пролиферация регуляторных клеток, что приводит к иммуносупрессии. Иммуносупрессия, вызванная сепсисом, обусловлена как врожденными, так и приобретенными иммунными дисфункциями и характеризуется высвобождением противовоспалительных цитокинов, гибелью иммунных клеток, истощением Т-клеток и избыточной генерацией иммунных регуляторных клеток, включая регуляторные Т-клетки (Tregs) и миелоидные супрессорные клетки (MDSCs).
При сепсисе иммуносупрессия тесно связана с клеточной анергией, толерантностью к эндотоксинам или истощением иммунных клеток. Снижение экспрессии человеческого лейкоцитарного антигена-DR (HLA-DR) и повышение уровня молекул иммунных контрольных точек, таких как белок программируемой клеточной смерти 1 (PD-1), Т-клеточный иммуноглобулин и муцин-домен-содержащий белок-3 (TIM-3), а также аттенюатор В- и Т-лимфоцитов (BTLA), еще более усугубляют иммуносупрессию.
Метаболические изменения являются важной движущей силой иммуносупрессии при инвазии. Исследования метаболизма Т-клеток у септических пациентов выявили изменения в механизме mTOR, что приводит к неспособности индуцировать гликирование, окислительное фосфорилирование и производство АТФ. В результате Т-клетки не получают достаточного количества энергии, что нарушает не только их функциональность, но и снижает их пролиферативную способность. Кроме того, толерантность к эндотоксинам считается одним из механизмов иммуносупрессии при сепсисе. Под толерантностью к эндотоксинам понимается сниженная реактивность клеток на стимуляцию эндотоксинами (липополисахаридами) после предварительного воздействия эндотоксинов.
Основные признаки толерантности к эндотоксинам включают снижение уровня воспалительных медиаторов, таких как фактор некроза опухоли-альфа (TNF-α), интерлейкин-1β (IL-1β), C-X-C мотив хемокина 10 (CXCL10), и повышение уровня противовоспалительных цитокинов, таких как IL-10 и трансформирующий фактор роста-бета (TGF-β). Поэтому толерантность к эндотоксинам часто рассматривается как регуляторный механизм хозяина против чрезмерного воспаления, имеющий терапевтическое значение. Когда септические пациенты переходят в фазу иммуносупрессии, происходят нарушения функциональности иммунных клеток, что приводит к быстрому ухудшению их состояния и значительному повышению уровня смертности.
3. Роль иммунных клеток при сепсисе
3.1 Роль нейтрофилов при сепсисе
На ранних стадиях инфекции немедленно активируется врожденная иммунная система, при этом нейтрофилы являются первичными фагоцитами, мигрирующими из кровотока в очаг инфекции. Нейтрофилы мигрируют к месту инфекции, направляемые сигналами от рецепторов и хемотаксических факторов, и эффективно поглощают и уничтожают патогены, выделяя реактивные формы кислорода, антимикробные белки и образуя внеклеточные нейтрофильные ловушки (NETs). Кроме того, они могут выделять медиаторы воспаления для усиления иммунного ответа.
В контексте сепсиса нейтрофилы играют особенно важную роль. В это время в кровотоке наблюдается значительный приток нейтрофилов, сопровождающийся подавлением апоптоза и увеличением периода полураспада, что приводит к повышению количества нейтрофилов. Микроорганизмы и продукты их жизнедеятельности, попадающие в кровоток, могут стимулировать значительное и быстрое увеличение количества нейтрофилов периферической крови, что может привести к истощению пула нейтрофилов костного мозга и выбросу незрелых клеток в кровь.
Незрелые нейтрофилы обладают низкой распознающей и фагоцитарной способностью, не могут эффективно удалять патогены, а их плохая пластичность способствует скоплению в капиллярах, вызывая окклюзию сосудов, гипоксию тканей и повреждение органов. Такие цитокины, как TNF-α, IL-1β, IL-6, IL-17, и бактериальные компоненты могут активировать G-CSF, способствуя дифференцировке нейтрофилов. Ингибируя сигнальную ось CXCR4/CXCL12, G-CSF может способствовать пролиферации и дифференцировке CD34+ миелоидных предшественников и миграции зрелых нейтрофилов из костного мозга. Более того, у пациентов с тяжелым сепсисом значительное повышение Mcl-1 может подавлять апоптоз нейтрофилов и способствовать увеличению продолжительности их жизни в несколько раз.
Бактериальный липополисахарид (ЛПС) и комплемент 5а (С5а) также могут продлевать жизнь нейтрофилов по следующим механизмам. Поскольку ЛПС и C5a активируют ERK1/2, PI3K и нижележащие пути Akt в нейтрофилах, фосфорилирование Bad ингибирует высвобождение митохондриального цитохрома C и уменьшает апоптоз. Кроме того, C5a может снижать апоптоз нейтрофилов за счет увеличения экспрессии Bcl-XL и снижения экспрессии Bim. Также ЛПС может ингибировать миграцию и расщеплять MNDA, тем самым предотвращая деградацию Mcl-1 под действием протеасом. Длительная жизнь нейтрофилов может позволить им выполнять более сложные функции в тканях, такие как купирование воспаления или запуск адаптивных иммунных реакций, но их постоянное присутствие в тканях может также вызвать повреждение тканей и органов.
Активированные нейтрофилы вырабатывают большое количество бактерицидных веществ. В борьбе с инфекциями взаимодействует целый ряд сложных физиологических механизмов, что приводит к иммунным нарушениям и системным воспалительным реакциям, а в дальнейшем вызывает дисфункцию свертывания крови и повреждение тканей. Активированные нейтрофилы инфильтрируют и накапливаются в важных органах, производя большое количество реактивных форм кислорода (ROS) через респираторный взрыв и высвобождая бактерицидные вещества через дегрануляцию, прямо или косвенно вызывая повреждение важных тканей и органов.
Генерация свободных радикалов кислорода, образующихся при респираторном взрыве, приводит к дисфункции митохондриальной системы трансмембранного транспорта веществ, и самое важное - к образованию кальциевой перегрузки. А кальциевая перегрузка разрушает устойчивое состояние концентрации Ca2+ внутри и вне клетки, что приводит к накоплению большого количества Ca2+ в митохондриях, делая клетки неспособными поддерживать нормальную функцию, приводя к дисфункции митохондрий, клеток и тканей.
Кроме того, многие провоспалительные и противовоспалительные факторы высвобождают индуцированные ROS, что приводит к дисбалансу окислительно-восстановительных состояний в организме, вызывая реакции окислительного стресса и в конечном итоге приводя к нарушению работы органов. Повышенное производство ROS может также нарушать функцию эндотелиальных клеток сосудов во всем организме, увеличивать проницаемость сосудов, нарушать функцию митохондрий и в конечном итоге вызывать дисфункцию органов и систем у пациентов с сепсисом.
Нейтрофилы также могут захватывать и очищать нефагоцитированные патогены благодаря выработке NETs. NETs - это уникальная форма клеточной смерти нейтрофилов, при которой они высвобождают волокна ДНК и гранулы, содержащие антимикробные белки, образуя паутинообразную структуру, которая захватывает и уничтожает патогены. На ранней стадии сепсиса NETs могут образовывать физический барьер, способствующий захвату и уничтожению патогенов, предотвращая их распространение и препятствуя прогрессированию сепсиса. Однако по мере прогрессирования заболевания NETs могут вызывать повреждение тканей, усиление аутоиммунитета и образование тромбов в кровеносных сосудах. Это может быть связано с тем, что NETs действуют как повреждающие молекулярные паттерны (DAMPs), активируя рецептор TLR9 для запуска воспалительного ответа, способствуя инфильтрации воспалительных клеток в ткани или органы и усугубляя повреждение тканей.
Гистоны - важные антимикробные компоненты NETs, которые также могут быть цитотоксичными для эндотелиальных клеток, вызывая повреждение эндотелиальных клеток и нарушая перфузию микрососудов. Они также могут способствовать выработке тромбина, активировать тромбоциты и ингибировать антикоагулянты, тем самым способствуя диссеминированному внутрисосудистому свертыванию крови и образованию тромбозов. Нейтрофилы через ось NET-тромбоциты-тромбин способствуют повышенной продукции NETs, что приводит к увеличению заболеваемости и смертности при сепсисе.
Кроме того, чрезмерная активация инфламмасом через каспазу-1-зависимый классический процесс пироптоза индуцирует гибель клеток, вызывая массивный выброс IL-1β и IL-18. Нейтрофилы проникают в неспецифические органы, включая печень и почки, вызывая высвобождение медиаторов воспаления из поврежденных клеточных мембран, тем самым усиливая воспалительный ответ, ускоряя прогрессирование сепсиса и вызывая тяжелое повреждение тканей и дисфункцию органов. В терминальной фазе сепсиса нейтрофилы истощаются, их миграция и функция нарушаются, поэтому нейтрофилы не могут достичь очага инфекции, чтобы контролировать инфекцию, но накапливаются в важных органах и вызывают серьезные повреждения.
ICAM-1 - молекула адгезии, экспрессируемая на эндотелиальных клетках сосудов, связывается с β2-интегринами, индуцированными на поверхности нейтрофилов, и является ключевой молекулой, опосредующей адгезию нейтрофилов. При сепсисе сродство между ICAM-1 и β2 интегринами повышается, что приводит к ригидности клеток, вызывает окклюзию сосудов и гипоксическое повреждение тканей, что является важным фактором органной недостаточности. Сверхэкспрессия ICAM в эндотелиальных клетках неспецифических органов также может быть основной причиной повреждения тканей органов и функциональных нарушений, вызванных сепсисом. Ненормальная аутофагия и пироптоз могут способствовать формированию NETs, вызывать повреждение мембраны нейтрофилов и высвобождение многочисленных провоспалительных цитокинов, а также еще больше усиливать воспалительный ответ. Кроме того, циркуляция незрелых нейтрофилов и появление супрессивных субпопуляций не только способствуют эффективному уничтожению инфекции, но и препятствуют активации и эффективности других клеток, таких как лимфоциты, способствуя тем самым развитию последующей иммуносупрессии.
3.2 Роль макрофагов при сепсисе
Макрофаги - важнейшие клетки врожденного иммунитета и антигенпрезентирующие клетки, обладающие высокой пластичностью. С одной стороны, они инициируют врожденный иммунный ответ, распознавая факторы риска в микроокружении; с другой стороны, они модулируют иммунный ответ хозяина посредством дифференциальной поляризации, формируя многомерный фенотипический спектр в ответ на изменения микроокружения. Таким образом, макрофаги играют важную роль в регуляции иммунного баланса хозяина и воспалительных реакций при сепсисе.
Основные известные фенотипы - это воспалительные или классически активированные (M1-подобные) макрофаги и заживляющие или альтернативно активированные (M2-подобные) макрофаги. Каждое из этих поляризованных состояний макрофагов имеет свои функции и только когда они находятся в равновесии, поддерживается иммунный гомеостаз хозяина.
На ранних стадиях сепсиса М1-подобные макрофаги могут быть активированы отдельными Th1-цитокинами (TNF-α и IFN-γ) или патоген-ассоциированными молекулярными паттернами (такими как ЛПС). Последние исследования показывают, что Caveolin-1 и окисленный липопротеин низкой плотности также играют важную роль в поляризации М1-подобных макрофагов. М1-подобные макрофаги высоко экспрессируют CD68, CD80, CD86, основной комплекс гистосовместимости (MHC)-II, индуцибельную синтазу оксида азота (iNOS) и Toll-подобный рецептор (TLR) 4. Они повышают экспрессию MHC-II, связываясь с ко-стимулирующими молекулами (CD80 и CD86) и способствуя развитию цитотоксического адаптивного иммунитета. Высокий уровень iNOS в M1-подобных макрофагах способствует синтезу оксида азота.
М1-подобные макрофаги выделяют большое количество хемокинов (CCL5 и CXCL5) для привлечения естественных клеток-киллеров, нейтрофилов и активированных Т-клеток. Кроме того, M1-подобные макрофаги вырабатывают значительное количество провоспалительных цитокинов (IL-6, IL-12, IL-23 и IL-1β), промежуточных продуктов реактивного кислорода и промежуточных продуктов реактивного азота для уничтожения патогенов хозяина. В целом М1-подобные макрофаги проявляют мощную цитотоксическую активность, способны уничтожать патогены, очищать аберрантные эндогенные ткани и клетки в иммунном микроокружении, способствовать деградации матрикса и проявлять противоопухолевую активность. Однако длительная поляризация M1-подобных макрофагов или ее усиление могут привести к повреждению тканей, органов и иммунных клеток.
Напротив, на поздней стадии сепсиса М2-подобные макрофаги могут быть активированы Th2-цитокинами (IL-4 и IL-13), TGF-β, IL-10, глюкокортикоидами и иммунными комплексами. М2-подобные макрофаги экспрессируют высокий уровень лектина С-типа (CD206) и рецептора скавенджера (CD163). Они способствуют секреции хемокинов (CCL17 и CCL18) для привлечения эозинофилов, базофилов, Th2 и регуляторных Т-клеток, демонстрируя противовоспалительный спектр цитокинов, вырабатывая высокие уровни IL-10, резистин-подобного альфа (Fizz1), антагониста рецептора IL-1 и TGF-β. Таким образом, М2-подобные макрофаги участвуют в иммунной регуляции, способствуют ангиогенезу, ремоделированию тканей и подавлению воспаления. Соответственно, целенаправленная модуляция поляризации и фенотипических изменений макрофагов в качестве адаптации к микроокружению может стать эффективным терапевтическим подходом к лечению сепсиса.
3.3 Роль Т-клеток при сепсисе
Клетки врожденного иммунитета, помимо своей роли в фагоцитозе и клиренсе патогенов, могут также перерабатывать патогены для выработки специфических антигенов и вызывать адаптивные иммунные реакции. Т-лимфоциты, в частности, занимают ключевую позицию в адаптивном иммунном ответе. Зрелые Т-лимфоциты, попадая в кровь, распознают антигены из основных молекул комплекса гистосовместимости с помощью поверхностных антител. После активации и пролиферации они выполняют свои биологические функции по уничтожению большинства патогенов. Клетки TH1 усиливают фагоцитарную активность и бактерицидную способность макрофагов, выделяя такие цитокины, как IFN-γ и TNF-α, способствующие развитию воспаления и клеточного иммунного ответа.
CD8+ Т-клетки идентифицируют и уничтожают инфицированные клетки. Регуляторные Т-клетки CD4+, CD25+ модулируют иммунный ответ, подавляют воспалительные реакции или предотвращают повреждение иммунной системой собственных тканей. Однако чрезмерная активация Т-лимфоцитов может привести к высвобождению значительного количества провоспалительных медиаторов, таких как IFN-γ, вызывая чрезмерное воспаление и усугубляя повреждение тканей и органов. Чрезмерное воспаление запускает противовоспалительные механизмы, приводящие к выработке противовоспалительных медиаторов, таких как TGF-β и IL-10, чтобы сбалансировать воспалительный ответ, что приводит к иммунной супрессии, характеризующейся апоптозом и функциональным подавлением лимфоцитов.
При прогрессировании сепсиса иммунная функция пациента продолжает нарушаться, что приводит к глубокому подавлению иммунитета и переходу в состояние иммунного паралича. Это может вызвать устойчивую дисфункцию органов и привести к рецидивирующим инфекциям, вплоть до угрожающих жизни состояний. Исследования показали, что истощение Т-лимфоцитов является основной характеристикой иммунной супрессии при сепсисе, и существует несколько механизмов, лежащих в основе истощения Т-лимфоцитов при сепсисе. Первый механизм включает сверхэкспрессию отрицательных ко-стимулирующих факторов клеточной поверхности, таких как PD-1, BTLA, CTLA-4, Tim-3 и LAG-3. Эти факторы подавляют активацию, пролиферацию или индукцию апоптоза в Т-лимфоцитах, что приводит к истощению Т-лимфоцитов. Уровень экспрессии отрицательных ко-стимулирующих факторов клеточной поверхности напрямую связан с тяжестью сепсиса.
Второй механизм связан с увеличением доли Tregs. Tregs оказывают иммуномодулирующее действие как во врожденном, так и в адаптивном иммунитете и могут индуцировать апоптоз в других лимфоцитах. Более того, экспрессия PD-1 на клетках Tregs напрямую связана с тяжестью сепсиса. Кроме того, активация гипоталамо-гипофизарно-надпочечниковой оси приводит к повышению концентрации кортизола, который связывается с глюкокортикоидными рецепторами и оказывает противовоспалительное действие. Это приводит к снижению активности лимфоцитов и их апоптозу. Стимуляция симпатической нервной системы вызывает повышение уровня катехоламинов, активацию бета-адренергических рецепторов, повышение секреции IL-10, снижение секреции TNF-α, повышение высвобождения противовоспалительных агентов, снижение высвобождения провоспалительных агентов, а также снижение активности Т-лимфоцитов, пролиферации и апоптоза.
Другие факторы, такие как повышенная экспрессия CaSR, также связаны с апоптозом Т-лимфоцитов при сепсисе. IL-7 играет важнейшую роль в пролиферации и созревании лимфоцитов. Исследования показали, что уровень IL-7 снижается у пациентов с сепсисом, что приводит к уменьшению количества Т-лимфоцитов, причем степень снижения коррелирует с тяжестью сепсиса. Таким образом, понимание влияния Т-лимфоцитов на иммунную функцию пациентов с сепсисом и мониторинг изменений Т-лимфоцитов может обеспечить лучшее понимание иммунного статуса пациента и эффективно направлять клинические вмешательства.
Другие иммунные клетки также вносят свой вклад в иммунную защиту организма. Например, дендритные клетки захватывают и перерабатывают патогены и презентируют их поверхностные антигены Т-лимфоцитам. В-клетки способствуют выработке плазматическими клетками антител для нейтрализации внеклеточных токсинов. Помимо этого, взаимодействие между различными иммунными клетками является важнейшим элементом иммунного ответа, включая взаимодействия, подобные тем, что происходят между нейтрофилами и Т-клетками. Помимо презентации антигенов, нейтрофилы также оказывают регулирующее воздействие на лимфоциты. При сепсисе IFN-γ может побуждать нейтрофилы экспрессировать PD-L1 и через сигнальный путь PD-L1 негативно регулировать лимфоциты, ингибировать их пролиферацию, активацию и высвобождение воспалительных цитокинов, а также способствовать апоптозу лимфоцитов. Он также может связываться с CD80, конкурентно препятствуя взаимодействию между CD80 и его лигандами, и, следовательно, препятствуя пути активации Т-клеток.
При сепсисе нейтрофилы также могут влиять на нормальный клеточный цикл Т-клеток, секретируя аргиназу-1, расщепляющую L-аргинин, в результате чего Т-клетки остаются в цикле G0-G1, что приводит к их дисфункции. Более того, некоторые субпопуляции нейтрофилов могут препятствовать пролиферации Т-клеток через Mac-1, подавлять высвобождение IFN-γ и препятствовать активности Т-клеток. В процессе воспаления Т-хелперы (Th) дифференцируются на Th1 и Th2 клетки, причем равновесие между этими двумя фракциями имеет решающее значение для сохранения иммунного равновесия. IL-12 и IL-4, секретируемые нейтрофилами, побуждают наивные CD4+ Т-клетки дифференцироваться в подтипы Th1 и Th2, соответственно. При сепсисе количество IL-4, выделяемого Th2, больше, а баланс между Th1 и Th2 нарушается, что приводит к иммуносупрессивному состоянию. Т-лимфоциты также могут активировать и усиливать функцию нейтрофилов, секретируя цитокины, такие как IFN-γ, или подавлять активность нейтрофилов, секретируя TGF-β, IL-10. Эти механизмы взаимодействия и регуляции имеют решающее значение для поддержания иммунного баланса и предотвращения чрезмерной воспалительной реакции. При сепсисе дисбаланс иммунной системы может привести к безудержной воспалительной реакции и MODS, поэтому важно изучить механизмы взаимодействия и регуляции между иммунными клетками.
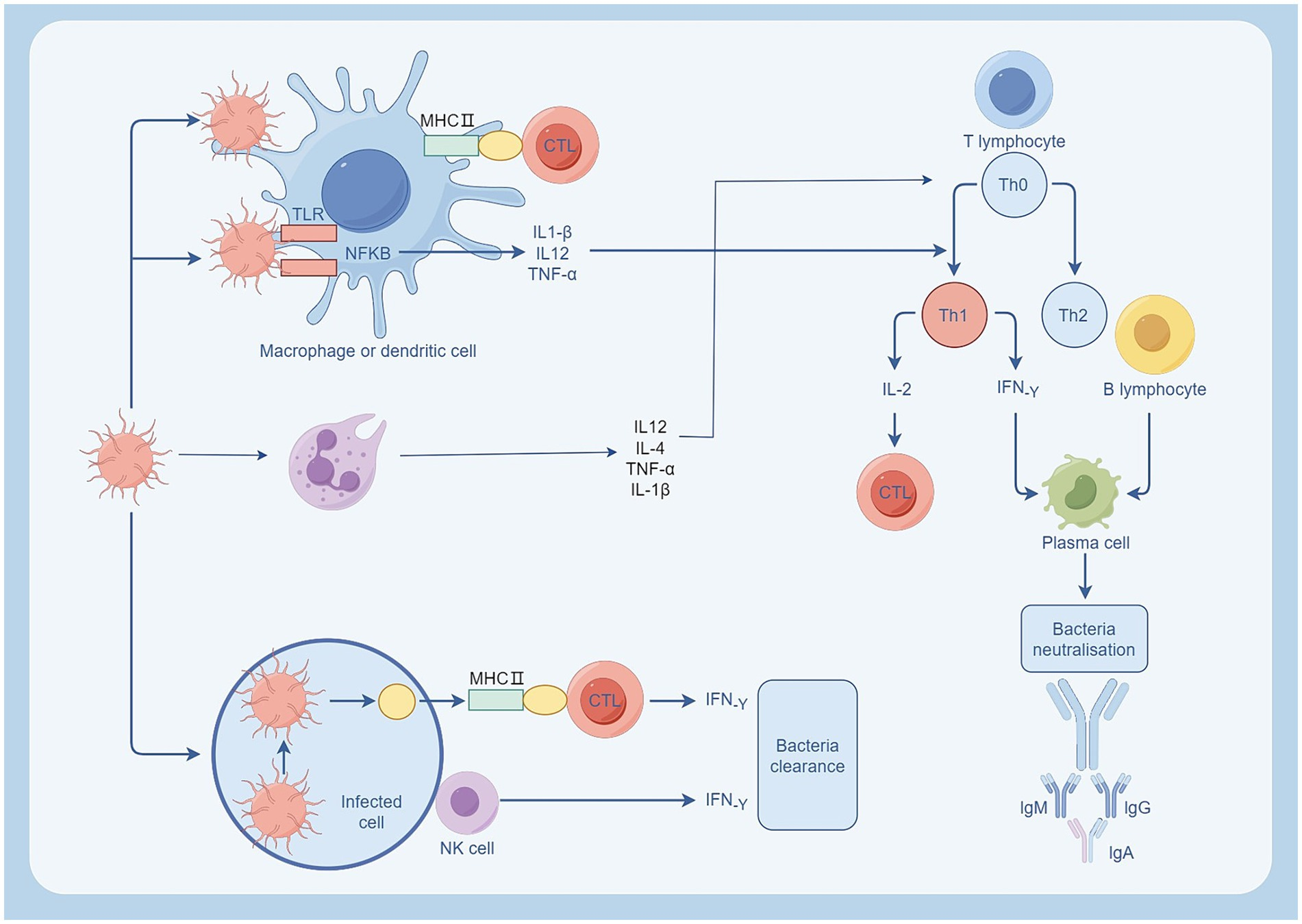
Ответ иммунных клеток на бактериальную инфекцию. Нейтрофилы и макрофаги активируются после бактериальной инфекции. Нейтрофилы регулируют Т-клетки, выделяя IL-2, IL-4, и способствуют развитию воспаления, выделяя TNF-α, IL-1β. Макрофаги поглощают патогены путем фагоцитоза и передают их Т-клеткам через MHC II, способствуя дифференцировке TH0 в TH1. Клетки TH1 активируют CTL, секретируя IL-2, и способствуют выработке антител плазматическими клетками через секрецию IF-γ, нейтрализующего внеклеточные токсины. Источник: Figdraw.
4. Роль цитокинов при сепсисе
Цитокины представляют собой обширную группу относительно небольших белков (<40 кДа), играющих ключевую роль в клеточной сигнализации, которые вырабатываются и выбрасываются в основном для облегчения межклеточной коммуникации. Цитокины связываются со специфическими рецепторами на различных типах клеток, вызывая активацию, пролиферацию или миграцию клеток-мишеней. Их можно разделить на несколько групп, включая хемокины, интерлейкины, TNF, интерфероны и факторы роста. При сепсисе наиболее изученными цитокинами являются TNF-α и IL-1, которые могут активировать клетки-мишени и стимулировать выработку дополнительных цитокинов. К другим важным цитокинам при сепсисе относятся IL-6, IL-8, IL-12, IFN-α, G-CSF и IL-10.
TNF в основном вырабатывается макрофагами и лимфоцитами. Когда его концентрация достигает определенного порога, он нарушает баланс воспалительных реакций, что приводит к развитию сепсиса. Был достигнут значительный прогресс в понимании механизма, с помощью которого TNF вызывает сепсис, сфокусировавшись на его способности вызывать окислительное повреждение, аномальное распределение кальция в клетках и активацию каспаз. TNF-α способен запускать такие ферменты, как NADPH-оксидаза и синтаза оксида азота, что приводит к перепроизводству реактивных видов кислорода. Чрезмерное накопление этих свободных радикалов кислорода может привести к окислительному повреждению, которое может повредить клеточные мембраны.
Кроме того, TNF-α способен способствовать истощению внутриклеточных восстановителей, таких как глутатион и глутатионпероксидаза, что усиливает масштабы окислительного повреждения. TNF-α также может способствовать высвобождению ионов кальция и ингибировать их отток, что приводит к повышению уровня внутриклеточного кальция. Высокий уровень внутриклеточного кальция может привести к истощению запасов ионов кальция в эндоплазматическом ретикулуме, что приводит к стрессу эндоплазматического ретикулума и нарушениям сворачивания белков. Помимо этого, высокий уровень ионов кальция может активировать сигнальные пути воспаления, такие как фосфолипаза A2 и протеинкиназа C. Связывание TNF-α со своими рецепторами может активировать такие сигнальные пути, как NF-κB и MAPK, которые в конечном итоге активируют каспазу-8 и каспазу-3, запуская каскад апоптоза. Раннее вмешательство, направленное на блокирование сигнальных путей, ответственных за передачу воспаления, а также ингибирование и нейтрализацию TNF-α, имеет большое значение для лечения сепсиса в клинических условиях.
IL-1β, признанный цитокином-катаболином, относится к семейству IL-1, представленному 11 генами, и образуется после активации инфламмасомы, в частности NLRP3. IL-1β служит ключевым ранним цитокином в иммунной модуляции и воспалительных реакциях, преимущественно синтезируясь активированными моноцитами/макрофагами, и играет важную роль в повреждении тканей. Он обладает большим потенциалом в опосредовании патологического повреждения, например активирует Т-клетки, стимулирует выработку клетками PGI2, IL-1, IL-6, способствует росту В-клеток, индуцирует экспрессию молекул адгезии в эндотелиальных клетках, стимулирует выработку матриксных металлопротеиназ и активатора плазминогена в синовиальных клетках, индуцирует резорбцию костей и синтез белков острой фазы в печени.
Коллективное действие этих факторов усиливает системный воспалительный ответ, а исследования показали, что уровень IL-1β повышен у пациентов, не переживших сепсис, по сравнению с выжившими, что указывает на связь между повышенным уровнем IL-1β и исходом сепсиса. Изучение медиаторов воспаления всегда было важным патолого-физиологическим аспектом развития сепсиса, поскольку их уровни и изменения напрямую отражают возникновение, развитие и прогноз сепсиса. В последние годы роль цитокинов в патогенезе повреждений органов, вызванных сепсисом, привлекает все большее внимание исследователей. Таргетная терапия, направленная против воспалительных путей, обещает кардинально предотвратить возникновение и прогрессирование повреждений органов, вызванных сепсисом.
В некоторых клинических исследованиях предпринимались попытки лечения сепсиса путем блокирования определенных аспектов воспалительного ответа, таких как фактор некроза опухоли и интерлейкин-1, которые являются специфическими ингибирующими мишенями, но результаты часто были неудовлетворительными. Эти испытания были начаты на основе доклинических исследований, которые свидетельствовали об их эффективности. Три факта подтверждают идею подавления цитокинов. Во-первых, пациенты с повышенным уровнем цитокинов более предрасположены к летальному исходу. Во-вторых, экспериментальные животные модели показывают, что блокирование цитокинов может улучшить исход заболевания. В-третьих, введение очищенных рекомбинантных цитокинов приводит к повреждению органов и летальному исходу у экспериментальных животных.
С момента начала этих испытаний было обнаружено еще несколько аспектов воспалительного ответа, а потенциальными новыми мишенями стали интерлейкин-18 и HMG-1. Тем не менее, прежде чем приступать к новым клиническим испытаниям, необходимо тщательно проанализировать, почему предыдущие вмешательства оказались бесполезными. Концепция блокирования отдельных повышенных цитокинов может оказаться слишком упрощенной для решения сложных проблем сепсиса. По мере того как пациенты проходят различные стадии септического ответа, могут возникать подходящие интервалы для ингибирования нескольких цитокинов, в то время как в другое время более целесообразным может быть усиление иммунного ответа.
5. Механизмы повреждения эндотелиальных клеток
Основными проявлениями сепсиса являются гипотония, MODS и ДВС-синдром. Ключевым звеном патогенеза сепсиса является повреждение эндотелиальных клеток. При повреждении эндотелиальных клеток утечка жидкости из сосудов приводит к гипотонии, ишемия важных органов вызывает их дисфункцию, а активация факторов свертывания крови - к тромбозу. Нарушение межклеточных соединений эндотелия и деградация гликокаликса - ключевые особенности повреждения эндотелиальных клеток при сепсисе. Соединительные структуры между эндотелиальными клетками делятся на три типа: адгезивные соединения, герметичные соединения и гэп-соединения. Эти соединительные комплексы играют ключевую роль в поддержании целостности тканей, регулировании сосудистой проницаемости, облегчении экстравазации лейкоцитов и содействии ангиогенезу. Когда на эндотелиальные клетки воздействует большое количество медиаторов воспаления, нарушается работа эндотелиальных клеточных соединений и целостность сосудов.
Гликокаликс состоит из мембраносвязанных доменов, содержащих основные белки (такие как протеогликаны и гликопротеины, связанные с олигосахаридами) и белки плазмы (такие как альбумин и антикоагулянты). В физиологическом состоянии его структура и состав остаются неизменными. Однако под воздействием патологических факторов, таких как TNF-α, окисленный липопротеин, липополисахарид, ишемия/реперфузия, гипергликемия или воспалительная стимуляция, гликокаликс подвергается деградации и отмиранию. Исследования показали, что толщина и целостность гликокаликса эндотелиальных клеток уменьшается под воздействием липополисахарида и TNF-α. Во время прогрессирования сепсиса провоспалительные цитокины часто вызывают активацию тучных клеток, что приводит к их дегрануляции и последующему высвобождению цитокинов, гистамина, протеаз, гепариназ и других деградирующих элементов гликокаликса. Этот процесс повреждает эндотелиальный гликокаликс и изменяет проницаемость эндотелиальных клеток.
С другой стороны, отслаивание гликокаликса освобождает интегрины и селектины, что приводит к усилению адгезии и экссудации лейкоцитов, воспалению эндотелия и тканей, повышению проницаемости сосудов, в результате чего экссудат, альбумин и другие растворители попадают в межклеточное пространство, усугубляя отек тканей и снижая артериальное давление. Когда целостность стенки кровеносного сосуда нарушается и стимулируется различными микроорганизмами и их метаболитами, эндотоксинами, воспалительными цитокинами и комплементом, тканевой фактор (TF) может экспрессироваться и высвобождаться эндотелиальными клетками, нейтрофилами, моноцитами, эозинофилами и тромбоцитами. Попадая в кровоток, он активирует фактор VII и образует комплекс TF/VIIa. Этот комплекс впоследствии активирует фактор X, катализируя превращение протромбина в тромбин. Через развивающийся механизм положительной обратной связи формируется обширный микрососудистый тромбоз.
Во время воспаления организм задействует три важнейших антикоагулянтных пути: AT, APC и TFPI. В нормальных физиологических условиях t-PA и u-PA, выделяемые эндотелиальными клетками, служат основными движущими силами фибринолиза, превращая плазминоген в плазмин для разрушения и уничтожения фибриновых сгустков. Одновременно эндотелиальные клетки могут вырабатывать ингибитор активатора плазминогена-1. При сепсисе, хотя уровни t-PA и u-PA повышаются, TNF-α и IL-1 могут увеличивать экспрессию PAI-1, что приводит к общему подавлению фибринолиза. Кроме того, развитию сепсиса может способствовать разрушение других барьеров организма, таких как гематоэнцефалический барьер, перитонеальный барьер и другие. Повреждение эпителиальных клеток также может привести к сепсису. Эпителиальные клетки выстилают поверхности различных органов и слизистых оболочек в организме, таких как дыхательные пути, пищеварительный тракт и мочеполовые пути. Повреждение эпителиальных клеток может позволить патогенам проникнуть в организм, вызывая инфекцию и последующее прогрессирование сепсиса.
6. Ферроптоз при сепсисе
Ферроптоз - это внутриклеточный железозависимый процесс клеточной смерти, включающий в себя смертельные реакции пероксидации липидов, включающий в себя перегрузку железом, генерацию ROS и повышение уровня полиненасыщенных жирных кислот в фосфолипидах. Это приводит к потере целостности клеточной мембраны, нарушению сшивания липидов, влияющему на нормальную работу клеточной мембраны, и окислительному повреждению макромолекул и клеточных структур, что в конечном итоге приводит к гибели клетки.
Механизм может включать ингибирование клеточного поглощения цистеина, что приводит к снижению внутриклеточного глутатиона (GSH) и инактивации глутатионпероксидазы 4 (GPX4), вызывая дисбаланс в окислительно-восстановительной системе организма, накопление избыточного количества перекисей липидов и запуск клеточной смерти. Морфологические признаки включают повышенное содержание цитоплазматических и липидных перекисей, наличие в цитоплазме митохондрий меньшего размера, чем обычно, с уплотненной и повышенной плотностью мембраны, уменьшенными кристами и разрывом внешней мембраны митохондрий. Интенсивный стресс во время сепсиса может привести к нарушениям метаболизма ионов, липидов и энергии. Дисрегуляция гомеостаза железа в организме может привести к накоплению и аномальному распределению железа, что ведет к железозависимой гибели клеток.
В физиологических условиях избыток Fe2+ в клетках окисляется до Fe3+ и накапливается в виде ферритина. Однако во время сепсиса инфекция стимулирует повышение регуляции коактиватора ядерного рецептора 4 (NCOA4), который специфически распознает ферритин, инициирует аутофагию ферритина, высвобождая большое количество Fe3+, повышая внутриклеточную концентрацию свободного железа и способствуя железозависимой гибели клеток. У пациентов с сепсисом также может значительно повышаться уровень ROS за счет реакции Фентона, при которой ROS реагирует с полиненасыщенными жирными кислотами с образованием токсичных перекисей липидов, вызывающих железозависимую гибель клеток.
В физиологических условиях организм также вырабатывает ROS и другие оксиданты, которые быстро восстанавливаются до безвредных веществ с помощью редукционной системы организма. Однако во время сепсиса дисрегуляция иммунной системы приводит к дисбалансу в восстановительной системе, вызывая нарушения перекисного окисления липидов и провоцируя железозависимую гибель клеток. Кроме того, во время инфекции организм вырабатывает большое количество воспалительных факторов, таких как IL-6, который подавляет выработку гепцидина в печени, что приводит к повышению концентрации железа в крови.
Железо является важнейшим катализатором, который способствует реакциям окислительного стресса, генерируя большое количество реактивных видов кислорода. Чрезмерное производство реактивных видов кислорода может привести к обширной гибели клеток, что в свою очередь приведет к дисфункции органов и полиорганной недостаточности. Кроме того, гиперферритинемия может увеличивать выработку медиаторов воспаления и подавлять выработку противовоспалительных медиаторов, вызывая повреждение собственных тканей в результате чрезмерных воспалительных реакций.
Исследования показали значительную корреляцию между повышенным уровнем железа в сыворотке крови, маркерами инфекции, уровнем перекисного окисления липидов, а также увеличением долгосрочной смертности и распространенностью когнитивных нарушений у пациентов с сепсисом. Поэтому снижение внутриклеточного отложения железа, ослабление воспалительных реакций и уровня перекисного окисления липидов, а также блокирование сигнальных путей, связанных с железозависимой гибелью клеток, может дать новое представление о методах терапии сепсиса. Все больше данных свидетельствуют о том, что ионы железа играют решающую роль в противовоспалительных процессах и в развитии сепсиса, а эффекты препаратов, направленных на молекулы, связанные с железом, таких как ингибиторы ионов железа, при сепсисе постепенно подтверждаются.
7. Прогресс в лечении сепсиса
В настоящее время клиническое лечение сепсиса в основном включает в себя жидкостную реанимацию, раннюю антимикробную терапию и комплексную терапию, такую как вазопрессоры, глюкокортикоиды и антимикробные пептиды. В связи с критической ролью иммунной регуляции при сепсисе иммунотерапия имеет большие перспективы и достигла значительных успехов в области онкологии. Иммунотерапия сепсиса в основном включает в себя модуляторы цитокинов, ингибиторы иммунных контрольных точек и антиапоптозные агенты для стимулирования пролиферации иммунных клеток.
Исследования на животных показали многообещающие результаты для IL-7 и PD-L1, в то время как исследования GM-CSF и IFN-γ продолжаются. Цитокиновые модуляторы регулируют воспалительный ответ, стимулируя провоспалительные цитокины или ингибируя противовоспалительные цитокины. Липополисахарид в сочетании с IFN-γ может подавлять аутофагию макрофагов, способствовать активации макрофагов, облегчать клиренс бактерий и повышать выживаемость. Ингибиторы иммунных контрольных точек, таких как PD-1/PD-L1, цитотоксический Т-лимфоцит-ассоциированный антиген-4 (CTLA-4) и индоламин-2,3-диоксигеназа, хорошо зарекомендовали себя в противоопухолевой иммунотерапии и имеют большой потенциал для лечения сепсиса. Блокада PD-1/PD-L1 может восстановить функцию нейтрофилов, моноцитов, Т-клеток и натуральных клеток-киллеров (NK) при иммуносупрессии, вызванной сепсисом.
Путь CTLA-4 вовлечен в нейтрофил-опосредованную дисфункцию Т-клеток при сепсисе, а антитела CTLA-4 могут улучшить выживаемость и функцию Т-клеток у септических мышей. IL-7 индуцирует секрецию IFN-γ, способствует пролиферации Т-клеток и ингибирует апоптоз. GM-CSF и G-CSF повышают продукцию гранулоцитов и макрофагов, подавляют цитокиновый шторм и поддерживают физиологию легких, демонстрируя перспективность в качестве иммуномодуляторов при сепсисе с иммунным параличом. Поскольку сепсис быстро прогрессирует, следует отметить недостатки иммуномодулирующей терапии. Иммунотерапия опухолей может приводить к значительной иммунной токсичности, включая кожные реакции, эндокринные нарушения, поражение печени и почек, желудочно-кишечную токсичность, пневмонию, а также редкие неврологические и сердечные токсичности. Пациенты с сепсисом склонны к более серьезным побочным реакциям, когда у них развиваются эти токсические эффекты. Кроме того, использование антибиотиков для лечения сепсиса может снизить эффективность иммуномодуляторов, что требует дальнейших исследований по совершенствованию иммунотерапии сепсиса.
Генная терапия рассматривается как один из наиболее перспективных новых методов лечения заболеваний. Выяснено множество сигнальных путей, участвующих в воспалительном каскаде сепсиса, в том числе NF-κB, JAK/STAT, PI3K/Akt/mTOR и p38/MAPK. Ингибирование сигнальных путей и экспрессии нижележащих генов стало развивающейся областью в лечении сепсиса. Однако многочисленные проблемы еще предстоит преодолеть.
Сигнальный путь NF-κB является классическим путем для изучения патогенеза сепсиса. NF-κB служит ключевым медиатором воспалительного ответа и имеет большое значение для развития сепсиса. В неактивном состоянии NF-κB может связываться с ингибирующими субъединицами белка-ингибитора κB (IκB) в цитоплазме, освобождая тем самым киназный комплекс IκB, который препятствует связыванию NF-κB с ядерными рецепторами и, следовательно, препятствует его миграции в ядро. Тем не менее, при стимуляции киназный комплекс IκB активируется, фосфорилируя определенные сайты и снимая ограничения с NF-κB, что способствует его транслокации в ядро и вызывает быстрый выброс таких цитокинов, как TNF-α, IL-1, IL-6, формируя воспалительный шторм.
Между тем, большое количество воспалительных цитокинов также может взаимодействовать с NF-κB, создавая петлю обратной связи. Кроме того, NF-κB может регулировать гены, связанные с апоптозом, в том числе ингибировать антиапоптотические факторы Bcl-2 и Bcl-xL и усиливать проапоптотические факторы Bax и Caspase. Чрезмерная активация пути NF-κB и массовый апоптоз макрофагов могут усугублять воспалительную реакцию и повреждение органов, что является ключевой причиной высокой смертности при сепсисе. Поэтому, воздействуя на путь NF-κB, регулируя активность иммунных клеток, таких как макрофаги, и снижая уровень цитокинов, управляемых NF-κB, таких как TNF-α и IL-1, можно эффективно ослабить воспалительную реакцию при сепсисе и снизить уровень смертности среди пациентов.
JAK/STAT-путь - сложный сигнальный каскад, который регулируется множеством факторов. Он играет важнейшую роль в патогенезе сепсиса, участвуя в передаче сигнала от различных цитокинов и формируя сетевой эффект. Благодаря своей уникальной силе и устойчивости регуляторные факторы могут воздействовать на JAK/STAT-путь с разных сторон и в разных целевых точках. JAK являются членами семейства растворимых тирозинкиназ Janus, связанных с рецепторами без собственной киназной активности. Семейство включает четыре представителя: JAK1, JAK2, JAK3 и Tyk2. JAK3 в основном ограничен гемопоэтическими клетками, в то время как JAK1, JAK2 и Tyk2 распространены более широко и участвуют в передаче сигнала от различных цитокинов и гормонов, таких как интерферон-7 (IFN-7), интерлейкин (IL) и факторы роста.
TNF-α и IL-6 служат не только маркерами для оценки тяжести и прогноза сепсиса, но и важнейшими медиаторами раннего воспаления, вызывающими дисфункцию органов. На ранней стадии инфекции каскадные реакции усиливаются высвобождением цитокинов, которые ускоряют острую фазу воспалительного ответа организма, приводят к адгезии нейтрофилов к эндотелиальным клеткам, активации системы коагуляции, что в конечном итоге вызывает развитие сепсиса. Активация STAT3 неразрывно связана с высвобождением IL-6 во время ответа острой фазы, вызванного воздействием эндотоксинов.
HMGB-1 - новый поздний медиатор воспаления, участвующий в патогенезе сепсиса. Он является важным фактором воспаления, вызывающим эндотоксин-индуцированную смерть, обладает широким спектром внеклеточных воспалительных эффектов и может ускорять дальнейшее развитие сепсиса, индуцируя и усиливая провоспалительные факторы. Исследования показали, что JAK/STAT-путь сильно активируется во время сепсиса, а экспрессия мРНК HMGB-1 в тканях значительно усиливается и демонстрирует устойчивую экспрессию. Поэтому ингибирование активации JAK/STAT-пути может значительно уменьшить каскадную реакцию воспалительного ответа после тяжелой инфекции.
Сигнальный путь PI3K/AKT на сегодняшний день является единственным известным сигнальным трансдукционным путем, ингибирующим аутофагию, что было подтверждено во многих исследованиях, связанных с опухолями и метаболическими заболеваниями. Существующие исследования также подтвердили, что этот путь участвует в регуляции экспрессии различных воспалительных факторов при сепсисе. PI3K - это липидная киназа, которая широко распространена в цитоплазме различных клеток млекопитающих. Получив сигналы от тирозинкиназ и рецепторов, связанных с G-белками, на поверхности клетки, она рекрутирует регуляторную субъединицу p85 в непосредственной близости от плазматической мембраны. После связывания с субъединицей p85 субъединица p110 катализирует превращение фосфатидилинозитол-4,5-бисфосфата (PIP2), субстрата внутри мембраны, в фосфатидилинозитол-3,4,5-трисфосфат (PIP3). Затем PIP3 связывается с N-концевым PH-доменом AKT, вызывая перемещение AKT из цитоплазмы в клеточную мембрану.
При участии PDK1 и PDK2 AKT фосфорилируется на сайте фосфорилирования треонина (Thr308) и сайте фосфорилирования серина (Ser473), что приводит к его активации. mTOR, серин/треониновая протеинкиназа, является эволюционно консервативной и активирует AKT. Активированная AKT может фосфорилировать mTOR, усиливая его активность. mTOR имеет как минимум две каталитические субъединицы в разных комплексах - mTORC1 и mTORC2. В настоящее время считается, что активированный mTOR оказывает аутофаг-регулирующее действие через два пути. Один из них - прямое фосфорилирование белков аутофагии, поскольку mTOR может фосфорилировать различные белки аутофагии, блокируя реакцию димеризации ULK1 и препятствуя образованию индуцированных аутофагосом, тем самым ингибируя аутофагию. Другая причина заключается в том, что mTOR служит точкой конвергенции для множества сигнальных путей. Он способен интегрировать сигналы от питательных веществ и факторов роста и регулировать жизненный цикл клетки, способствуя транскрипции и трансляции.
Вмешательства, направленные на различные компоненты пути аутофагии и модулирующие активность сигнальных путей на разных стадиях заболевания, могут стать новыми терапевтическими подходами и прорывами в лечении. mTOR - относительно легко регулируемая мишень, которая была обнаружена в опухолях и при изучении других заболеваний. Ингибирование mTOR может эффективно активировать аутофагию, и наоборот. В настоящее время ингибиторы сигнального пути PI3K-AKT-mTOR используются в основном для лечения рака, а для их применения в лечении сепсиса необходимы дополнительные исследования.
Семейство MAPK - это серин/треониновые протеинкиназы, существующие в большинстве клеток млекопитающих. Оно катализирует обратимое фосфорилирование белков и активирует каскадную киназную реакцию, имея высококонсервативную молекулярную структуру. Четыре изоформы семейства p38 MAPK активируются путем двойного фосфорилирования в сайтах треонина (T) и тирозина (Y) MAPK-киназами (MKKs). Эти два участка разделены одной аминокислотой и образуют петлю активации с мотивом TGY, которая активирует цитокины и регулирует такие физиологические процессы, как воспаление, апоптоз и окислительный стресс. Исследования показали, что путь p38 MAPK может регулировать выделение иммунными клетками провоспалительных цитокинов. Например, активированный p38 MAPK способствует тому, что моноциты выделяют IL-1 и TNF-α, а нейтрофилы - IL-8.
В цитоплазме активированная p38 MAPK способствует биосинтезу TNF-α, повышая экспрессию белков 2 и 3 MAPK-активируемых протеинкиназ. Во время сепсиса клетки тканей находятся в состоянии стресса, и путь p38 MAPK легко активируется медиаторами воспаления, тепловым шоком или реактивными видами кислорода (ROS). ROS может активировать нижележащие цитокины, соединяясь с Grb2, тем самым участвуя в активации сигнального пути p38 MAPK. p38 MAPK косвенно активирует H3, фосфорилируя MSK1 на нижнем уровне, а H3 участвует в формировании хроматина, связываясь с ДНК в ядре. Чрезмерное фосфорилирование H3 может вызывать конденсацию хроматина и остановку клеточного цикла, способствуя тем самым апоптозу. p38 MAPK также может приводить к накоплению p53 сверх определенного порога, запуская апоптоз путем фосфорилирования сайта Ser15 p53. Таким образом, путь p38 MAPK регулирует прогрессирование сепсиса путем модуляции окислительного стресса, высвобождения медиаторов воспаления и апоптоза. Поэтому ингибирование активности пути p38 MAPK может стать новым терапевтическим подходом к лечению сепсиса. Ингибиторы пути p38 MAPK в основном используются для лечения рака, а препараты для лечения сепсиса все еще находятся в стадии изучения.
Заключение
В данном обзоре описаны патофизиологические механизмы и стратегии лечения сепсиса, который до сих пор не был достаточно хорошо изучен, поскольку он действительно сложен и индивидуально сильно различается. Многочисленные исследования показали, что сепсис, как многофакторное заболевание, тесно связан с взаимодействием между иммунными клетками, шквалом воспалительных факторов, повреждением эндотелиальных клеток и ферроптозом. Молекулярная биология открывает интригующие перспективы для лечения сепсиса, позволяя препятствовать его прогрессированию путем воздействия на сигнальные пути, задействованные в воспалительном каскаде. Однако исследователям крайне важно лучше понять сложную взаимосвязь между этими фундаментальными механизмами и характеристиками. Поэтому необходимы постоянные исследования для выяснения взаимосвязи между первопричинами, провоцирующими факторами и клиническим лечением сепсиса, что позволит разработать новые терапевтические концепции, имеющие большую научную и клиническую ценность.









