


За последние несколько десятилетий использование технологий секвенирования следующего поколения (NGS) в клинической диагностике расширилось.
Технологии NGS (next generation sequencing) позволяют проводить массовое параллельное секвенирование миллионов фрагментов ДНК с высокой пропускной способностью. В области диагностики генетических заболеваний и рака эти технологии стали золотым стандартом диагностики. Только в последнее десятилетие, когда эти технологии NGS стали более доступными, их использование в лаборатории клинической микробиологии стало реальностью.
Современные технологии NGS могут быть использованы в трех различных областях клинической микробиологической лаборатории:
- секвенирование всего генома (WGS- whole genome sequencing),
- целевое секвенирование метагеномов и
- метагеномное секвенирование методом "дробовика".
В данном обзоре мы проанализируем эти три области применения и представим исчерпывающую информацию о том, как они используются в настоящее время в здравоохранении, фундаментальных исследованиях и лабораториях клинической микробиологии. Эти приложения нелегко внедрить в лабораторию, поэтому мы подчеркиваем важные факторы, которые необходимо учитывать при внедрении этих технологий. Тем не менее, эти технологии уже изменили ландшафт обычной лаборатории клинической микробиологии, и мы обсуждаем, что, по нашему мнению, возможно для этих технологий в диагностике инфекционных заболеваний в ближайшие 5 лет.(Статья опубликована в 2022 г. - прим.пер.).
Эволюция технологий секвенирования
За последние 60 лет технологии секвенирования стремительно развивались, начав с секвенирования первого поколения и развившись до современных технологий секвенирования третьего поколения (табл. 1). Зарождение секвенирования первого поколения произошло одновременно, когда Фредерик Сэнгер и Аллан Максам и Уолтер Гилберт опубликовали свои протоколы, описывающие способы секвенирования ДНК.
Метод секвенирования Максама-Гилберта был основан на химическом разрушении, когда радиомеченную ДНК обрабатывали различными химическими веществами, чтобы разорвать цепь на определенных основаниях. Затем эти фрагменты прогоняли в полиакриламидном геле, чтобы определить положение интересующего нуклеотида. Сэнгер предложил метод обрыва цепи, который предполагает использование химических аналогов дезоксинуклеотидов (dNTPs), известных как дидезоксинуклеотиды (ddNTPs), которые предотвращают дальнейшее удлинение цепи ДНК. Метод включает четыре параллельные реакции с ddNTPs, которые проводятся в полиакриламидном геле для интерпретации того, какое основание присутствует в нуклеотидной последовательности.
Именно усовершенствования метода Сэнгера, такие как переход на флуорометрическую детекцию и детекцию с помощью капиллярного электрофореза, позволили продвинуть секвенирование ДНК до автоматизированных приборов для секвенирования ДНК.
Второе поколение технологий секвенирования появилось, когда вместо радиомеченых или флуоресцентно меченых нуклеотидов исследователи стали измерять с помощью люминесценции синтез пирофосфата для определения последовательности нуклеотидов. Этот метод, известный как "пиросеквенирование", и приборы, созданные для проведения пиросеквенирования, позволили проводить параллельные реакции секвенирования, что увеличило количество ДНК, которое можно секвенировать за один прогон.
Развивались и другие методы параллельного секвенирования, включая секвенирование с помощью системы лигирования и детекции олигонуклеотидов (SOLiD, Applied Biosystems), которое секвенирует ДНК путем лигирования, и метод секвенирования Ion Torrent, который измеряет разницу в рН, вызванную высвобождением протонов при полимеризации. Наиболее заметным и успешным из этих методов секвенирования второго поколения является метод секвенирования с мостовой амплификацией, который первоначально применялся компанией Solexa, а затем был приобретен компанией Illumina. В этом методе фрагментированная ДНК, меченная адаптером, проходит над участком комплиментарных олигонуклеотидов, закрепленных на проточной кювете. После связывания твердофазная полимеразная цепная реакция (ПЦР) создает кластеры клональных популяций из каждой отдельной исходной нити ДНК, связанной с проточной кюветой. Первый из этих приборов Illumina Genomic Analyzer был способен производить только очень короткие чтения, но он генерировал парные данные с последовательностью на каждом конце. Эти парные данные предоставляют больший объем информации, что обеспечивает большую точность при меньших фоновых помехах по сравнению с другими методами второго поколения.
До сих пор нет единого мнения о том, что определяет разницу между технологиями секвенирования второго и третьего поколения, но для целей данного обзора мы определяем секвенирование третьего поколения как возможность проведения секвенирования одной молекулы. Технологии одномолекулярного секвенирования не требуют этапов амплификации ДНК, что позволяет повысить пропускную способность, ускорить время выполнения теста и увеличить длину чтения.
Несколько компаний стали лидерами в разработке технологий секвенирования третьего поколения, и две наиболее заметные из них - Pacific Biosciences (PacBio, США) и Oxford Nanopore Technologies (Великобритания). Метод PacBio измеряет включение ДНК-полимеразой флуоресцентно меченых нуклеотидов в шаблон комплементарной последовательности. В центре этой технологии находится плотный массив наноструктур с нулевой длиной волны (ZMW - zero-mode wavelength). Эти ZMW-наноструктуры позволяют измерять отдельные флуоресцентно меченные нуклеотиды в режиме реального времени и за короткий промежуток времени. Эта технология способна производить очень длинные чтения длиной до 10 килобаз (кб). Метод Oxford Nanopore Technologies использует нанопоры, как биологические, так и твердотельные, встроенные в мембрану с ионным током. Одноцепочечная геномная ДНК или РНК может проходить через нанопоры, и каждое отдельное нуклеотидное основание физически блокирует ток, который может быть измерен с помощью стандартных электрофизиологических методов.
Что касается современной клинической микробиологической диагностики, то для идентификации бактериальных и грибковых видов по прямому росту колоний часто используется технология секвенирования по Сэнгеру первого поколения. Для идентификации бактерий используются гены 16S рибосомальной РНК (рРНК) и β-субъединицы ДНК-зависимой РНК-полимеразы (rpoB), а для грибов - гены рибосомального внутреннего транскрибируемого спейсера (ITS - internal transcribed spacer) и 28S рРНК.
В настоящее время новые поколения секвенирования (второе и третье) изучаются для использования в клинической микробиологии для идентификации интересующих патогенных организмов. Существует два основных способа достижения этой цели: первый - это улучшение идентификации растущих в культуре организмов с помощью секвенирования всего генома (WGS - whole-genome sequencing). Второй - использование метагеномного секвенирования для идентификации потенциальных патогенных организмов непосредственно из источника, чтобы обойти процесс культивирования и/или улучшить результат. В данном обзоре будут рассмотрены оба эти направления.
3. Полногеномное секвенирование микроорганизмов
Секвенирование всего генома (WGS) - это процесс секвенирования и сборки микробного генома интересующего организма. Эти микробные геномы могут принадлежать бактериям, грибам и вирусам.
WGS для бактерий, микобактерий и грибковых организмов требует культивирования и выделения организма перед выделением нуклеиновых кислот и последующим построением последовательности.
Это является ограничением для организмов, которые трудно или невозможно вырастить в культуре. В случае вирусных геномов WGS используется путем секвенирования непосредственно образца в отношении интересующего вирусного генома, что будет рассмотрено далее в разделе о метагеномном секвенировании.
Для WGS используются технологии секвенирования второго поколения (например, Illumina) или третьего поколения (например, PacBio или Nanopore), преимущества и недостатки которых более подробно описаны в других источниках. Существует множество нюансов того, как получить чистую культуру организма от культуральной чашки до конечных результатов секвенирования с помощью технологий обоих поколений, но в целом рабочий процесс одинаков (рис. 1).
Вкратце, сначала организм извлекается из культуральной чашки и из него выделяется ДНК. После извлечения ДНК создается библиотека, в которой ДНК каждого отдельного организма разрезается на фрагменты и снабжается адаптерами, содержащими уникальный штрих-код, что позволяет проводить мультиплексирование сотен образцов. Эти индивидуальные библиотеки объединяются вместе и передаются в выбранную технологию NGS. После завершения секвенирования проводится биоинформатика для демультиплексирования образцов, затем выполняется фильтрация качества и удаление адаптеров.
Затем существует три способа сборки генома для получения идентификации с помощью WGS. Первый известен как референсная сборка, при которой фрагменты ДНК выравниваются по известному референсному геному и получается консенсусный геном, как пазл. Второй - сборка de novo, при которой все фрагменты ДНК собираются в контиги. (Контиг, от англ. contiguous, представляет собой набор перекрывающихся сегментов ДНК, которые в совокупности представляют собой консенсусную область ДНК. В задаче сборки генома контиги представляют собой продолжительные участки ДНК (строки из нуклеотидов), полученные в процессе сборки - прим.пер.). При сборке de novo трудно получить геномы высокого качества, поэтому более популярным третьим конвейером сборки является гибрид сборки de novo и эталонной сборки, когда фрагменты ДНК собираются в контиги, а затем эти контиги сопоставляются с эталонными геномами.
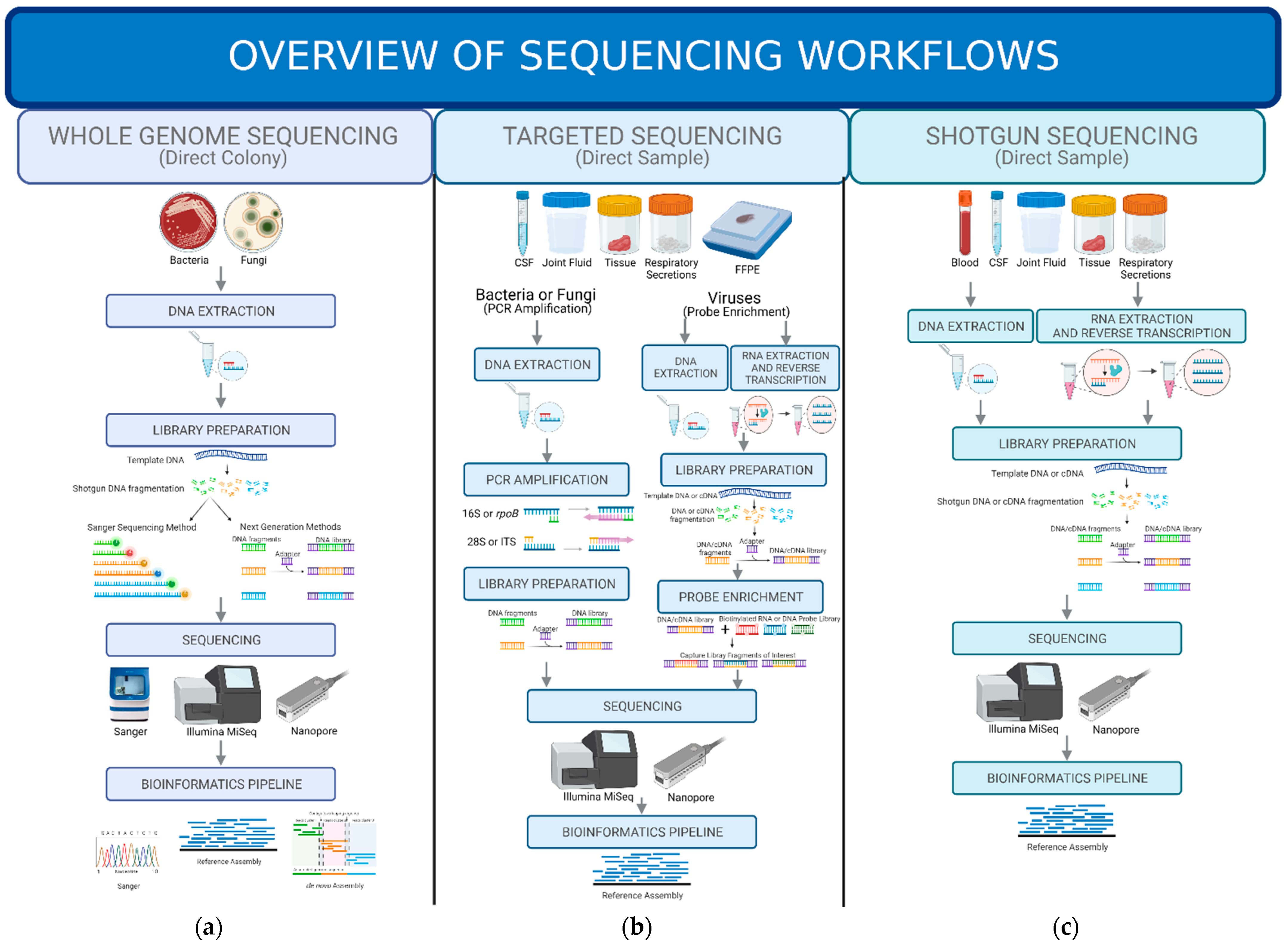
Рисунок 1. Обзор рабочих процессов секвенирования. (a) Полногеномное секвенирование - этот рабочий процесс начинается с получения колонии интересующего микроорганизма. Затем ДНК выделяется, фрагментируется и помещается в библиотеку для подготовки к секвенированию по методу Сэнгера или для других методов NGS. Затем библиотека секвенируется и анализируется с помощью биоинформационного конвейера. (b) Таргетное секвенирование - этот рабочий процесс начинается с клинического образца и включает в себя процесс отбора или обогащения перед подготовкой библиотеки в случае бактерий и грибов. Если интересующим патогеном является вирус, отбор или обогащение происходит после подготовки библиотеки. Затем подготовленные образцы секвенируются и анализируются с помощью биоинформационного конвейера. (c) Секвенирование по методу "дробовика" - этот процесс похож на процесс WGS, но вместо колонии интересующего микроорганизма ДНК или РНК выделяется непосредственно из представленного клинического образца. Выделенная ДНК или РНК проходит через подготовку библиотеки и затем секвенируется. Полученные результаты анализируются с помощью биоинформатики.
WGS микроорганизмов, которые можно культивировать и выделять, может быть использована для идентификации организма, типирования организма в эпидемиологических целях и выявления возможной резистентности организма к антимикробным препаратам. Традиционные методы клинической микробиологии для начальных этапов идентификации культивируемых бактерий включают основные морфологические наблюдения, биохимические тесты и идентификацию с помощью матрично-ассистированной лазерной десорбции-ионизации времяпролетной масс-спектрометрии (MALDI-TOF MS); последний метод по-прежнему очень точен и быстр по сравнению с WGS.
Однако бывают случаи, когда MALDI-TOF MS не позволяет уверенно определить видовую принадлежность, особенно в случае с прихотливыми организмами и анаэробными бактериями. В большинстве случаев достаточно идентификации до уровня рода, но есть случаи, когда видовая идентификация является обязательной из-за различий в профиле чувствительности к антибиотикам между видами.
Например, Enterococcus faecium и Enterococcus faecalis имеют два разных профиля чувствительности к антибиотикам, что имеет клиническое и эпидемиологическое значение. E. faecium обладает резистентностью к ампициллину, и этот вид энтерококков имеет высокий уровень резистентности к ванкомицину, в то время как у E. faecalis и то, и другое встречается относительно редко. С помощью MALDI-TOF MS удается очень точно идентифицировать эти два вида Enterococcus, но это всего лишь крайний пример, показывающий, что идентификация до рода не всегда является достаточной.
Современные методы микробиологической идентификации не позволяют определить серотип бактерии, что очень важно в случаях инфицирования сальмонеллами, когда для лечения очень важно выявить серотип возбудителя. В настоящее время в США идентификация серотипа сальмонелл с помощью WGS проводится в лабораториях общественного здравоохранения. До появления WGS лаборатории использовали импульсный полевой гель-электрофорез (PFGE - pulse-field gel electrophoresis) для идентификации серотипов Salmonella, но сравнительные исследования WGS показали, что он сопоставим с PFGE и WGS стал золотым стандартом.
Существует множество различных биоинформационных подходов, которые используются вместе с WGS для типирования конкретного изолята, и один из основных подходов известен как мультилокусное типирование последовательностей (MLST- multi-locus sequence typing). Этот подход использует набор "домашних" генов (5-7 генов в зависимости от бактерии) для конкретного вида бактерий и использует мутации в этих генах для сравнения степени родства одного бактериального изолята с другим.
MLST с выделением типов последовательностей (ST - sequence type) долгое время был золотым стандартом для типирования организмов с помощью WGS-анализа. Однако в 2014 году Leekitcharoenphon et al. использовали несколько типов биоинформационных анализов, чтобы сравнить различные методы типирования WGS с PFGE для кластеризации вспышек. Они сравнили 18 изолятов Salmonella enterica серовара Typhimurium из шести различных вспышек с помощью PFGE и четырех различных биоинформационных подхода к типированию и обнаружили, что анализ однонуклеотидных полиморфизмов (SNP - single nucleotide polymorphism) был методикой, способным кластеризовать 18 изолятов в соответствующие кластеры вспышек со 100% совпадением.
Хотя этот метод не является золотым стандартом, все больше лабораторий отдают предпочтение SNP-анализу для эпидемиологического отслеживания потенциальных вспышек, поскольку при таком анализе учитывается весь геном, а не только несколько отдельных генов.
В США результаты WGS, полученные лабораториями общественного здравоохранения, курируются и поддерживаются PulseNet, которая представляет собой сеть лабораторий общественного здравоохранения и агентств по контролю качества пищевых продуктов, координируемую Центрами по контролю заболеваний (CDC). Основная задача этой сети - помочь выявить и расследовать потенциальные вспышки в системах производства и распространения пищевых продуктов, что является основной инициативой подхода CDC, известного как One Health. Помимо вышеупомянутой идентификации серотипов Salmonella, PulseNet также проводит WGS для подтипирования и идентификации следующих микроорганизмов: Escherichia coli (O157 и другие E. coli, продуцирующие токсин Шига), Campylobacter, Listeria monocytogenes, Shigella, Vibrio cholerae, Vibrio parahaemolyticus и Cronobacter.
Идентификация серотипов при вспышках заболеваний пищевого происхождения - не единственная область применения WGS в здравоохранении. WGS также используется для эпиднадзора за заболеваниями, предотвращаемыми с помощью вакцин, такими как серогруппы Neisseria meningitidis, серотипы Streptococcus pneumoniae и патогены, резистентные к антимикробным препаратам, такие как Mycobacterium tuberculosis с множественной лекарственной устойчивостью. Одним из заболеваний, предупреждаемых вакцинацией, требующим постоянного мониторинга, является N. meningitidis, поскольку ежегодно происходят многочисленные вспышки. Возможность определить серогруппу и провести WGS изолятов N. meningitidis является ключевым фактором для оказания помощи в сфере общественного здравоохранения, чтобы определить, начинается ли вспышка и необходимы ли меры общественного здравоохранения, такие как массовая вакцинация.
В фундаментальных исследованиях WGS использовали для характеристики изолятов стрептококка группы В (GBS) серотипа IV, выделенных при инвазивных заболеваниях новорожденных, возникших в США и Канаде. Изоляты GBS серотипа IV имеют разнообразный генетический фон, включающий несколько различных ST и клональных комплексов (КК). Интерес представляет высоко распространенная линия ST-452, отнесенная к клональному комплексу CC23, описанному в нескольких странах. WGS и филогенетический анализ основного генома позволили предположить, что ST-452 могла возникнуть в результате генетической рекомбинации с исходным событием и штаммом-основателем, возникшим в результате переноса одного гена между CC23 и гипервирулентной линией CC17. Хромосомное картирование основных факторов вирулентности GBS показало, что штаммы ST-452 имеют уникальный профиль среди штаммов CC23 и CC17. Конъюгация и гомологичная рекомбинация с обменом крупными хромосомными фрагментами, охватывающими сотни килобаз, считаются одним из основных событий, контролирующих непрерывную эволюцию GBS. Резистентность к антимикробным препаратам у GBS также изучалась методом WGS с исследованием большого количества изолятов GBS, колонизирующих беременных женщин.
Большинство изолятов GBS серотипа IV в этом исследовании были ST-459, резистентными к тетрациклину, эритромицину и клиндамицину, впервые обнаруженными в Миннесоте, США, которые считаются основным фактором распространения GBS серотипа IV в Северной Америке. WGS-исследования рекомбинационных событий выявили многочисленные эпизоды капсульного перехода среди этих изолятов. Поразительное генетическое разнообразие и продолжающаяся эволюция GBS убедительно подтверждают необходимость текущего и будущего геномного мониторинга среди всех 10 серотипов GBS, поскольку полученная информация может оказать большое влияние на разработку вакцин против GBS.
В клинической лаборатории WGS доказал свою ценность в программах профилактики госпитальных инфекций, поскольку позволяет выявлять и отслеживать вспышки в пределах стационара.
В значительной части опубликованных работ WGS использовалась для отслеживания вспышек наиболее распространенных нозокомиальных патогенов - метициллин-резистентного золотистого стафилококка и Clostridiodes difficile. WGS помогла отследить вспышки таких тяжелых организмов с множественной лекарственной резистентностью, как резистентная к карбапенемам Klebsiella pneumoniae, резистентный к ванкомицину Enterococcus faecium и резистентная к множеству лекарственных препаратов Acinetobacter baumannii.
В больничных условиях бывают случаи, когда вспышки вызываются более прихотливыми организмами, и WGS используется для идентификации и эпидемиологического отслеживания этих прихотливых организмов, таких как Mycobacterium chimaera. Еще в середине 2010-х годов наблюдался рост заболеваемости инвазивными инфекциями M. chimaera у лиц, ранее перенесших операцию на открытой грудной клетке. Эти изоляты M. chimaera были секвенированы и оказались генетически и эпидемиологически связаны с контаминацией водонагревателей-охладителей, использовавшихся при кардиохирургических операциях у этих пациентов. Эти инфекции не были единичными случаями, поскольку такие водонагреватели-охладители были распространены по всему миру. В этих опубликованных случаях WGS проводился в ответ на подозрение о вспышке инфекции, но можно представить, что по мере повышения эффективности и рентабельности технологии госпитальные инфекционные программы смогут использовать эти технологии в своих диагностических микробиологических лабораториях для мониторинга и предотвращения будущих вспышек.
Как видно из нескольких примеров, приведенных выше, WGS может не только обеспечить идентификацию и эпидемиологическое отслеживание организмов, но и дать полный профиль присутствующих генов резистентности к противомикробным препаратам.
Современные методы выявления антимикробной резистентности - это либо фенотипические методы, зависящие от культуры, либо быстрые молекулярные методы, не зависящие от культуры. Культурно-зависимые фенотипические методы зависят от роста организма для интерпретации потенциальной резистентности, а культурно-независимые методы могут выявить только несколько основных маркеров генов резистентности.
Возможность получить полный генотипический профиль антимикробной резистентности организма позволяет получить более полный отчет о потенциальных механизмах антимикробной резистентности, присутствующих в организме. Существует множество опубликованных отчетов, которые показывают перспективность использования WGS для прогнозирования антимикробной резистентности для обычных микроорганизмов, таких как E. coli, S. aureus, Enterococcus faecium, Pseudomonas aeruginosa и Neisseria gonorrhoeae. Основной темой всех этих сообщений была способность генотипического прогнозирования резистентности с помощью WGS соответствовать фенотипической резистентности, зависящей от культуры, что является обязательным условием даже для рассмотрения возможности использования WGS вместо существующих методов. Однако современное состояние технологии WGS не позволяет полностью заменить существующие методы, а скорее дополнить их.
В настоящее время WGS занимает больше времени по сравнению с традиционными культурально-зависимыми и культурально-независимыми методами для обычных организмов, упомянутых выше. В случае, когда WGS может сразу же оказать влияние на прогнозирование резистентности к противомикробным препаратам, речь идет об организмах, которые долго выращиваются или для которых тестирование на чувствительность к противомикробным препаратам является трудоемким, как, например, для N. gonorrhoeae, о которых говорилось ранее, а также для видов Mycoplasma и Ureaplasma.
Еще одна группа труднорастущих организмов, где WGS может помочь ускорить выявление резистентности к противомикробным препаратам, - это медленно растущие микобактерии, такие как Mycobacterium tuberculosis с множественной лекарственной резистентностью. В одном крупном исследовании, известном как Comprehensive Resistance Prediction for Tuberculosis: An International Consortium (CRyPTIC), было проанализировано более 10 000 изолятов M. tuberculosis, и было установлено, что генотипический прогноз для препаратов первого ряда против туберкулеза коррелирует с фенотипической чувствительностью >90%.
Недавнее исследование подтвердило, что WGS может значительно сократить время выполнения традиционного анализа на чувствительность к противомикробным препаратам, который может занимать до месяца. Еще один профиль резистентности микобактерий к противомикробным препаратам, где WGS может быть полезен, - это Mycobacteroides abscessus (ранее Mycobacterium abscessus), которая может обладать индуцируемой резистентностью к кларитромицину, а золотым стандартом для выявления этой резистентности является инкубация бульонных микроразведений в течение 14 дней. Одна из групп подтвердила и внедрила WGS-тест, позволяющий предсказать эту индуцированную устойчивость к кларитромицину, а также резистентность к амикацину всего за 3-5 дней по сравнению со стандартом в 14 дней. Такие случаи уменьшения времени выполнения теста облегчат врачам лечение таких сложных случаев.
Одним из недостатков выявления резистентности к противомикробным препаратам с помощью WGS является то, что выявляются только известные гены резистентности и мутации. Однако продолжение секвенирования организмов с множественной лекарственной резистентностью может привести к открытию новых генов резистентности и механизмов резистентности. Более того, многие группы работают над созданием программ машинного обучения для поиска резистентности к противомикробным препаратам.
Использование WGS для идентификации грибов в клинической диагностике менее обосновано, чем для идентификации бактерий, но можно увидеть, где это может быть более выгодно по сравнению с существующими морфологическими методами идентификации в микологии, которые могут быть субъективными. В одном из исследований было показано, что корреляция между фенотипической идентификацией, использующей микроскопическую и колониальную морфологию и физиологические исследования, и секвенированием области D2 большой субъединицы гена рРНК и полных областей ITS многих распространенных и редких клинически значимых плесеней составляет приблизительно 50%.
WGS использовался в нескольких расследованиях вспышек контаминации лекарственных препаратов, например в Чили и Колумбии, где препарат против тошноты был контаминирован Sarocladium kiliense, который вызвал инфекции кровотока у людей. Другим примером вспышек грибковых заболеваний, вызванных контаминацией медикаментов, является Exserohilum rostratum. Более 13 000 пациентов получили различные виды инъекций (эпидуральные, параспинальные и суставные) контаминированным ацетатом метилпреднизолона (MPA). WGS удалось связать эти случаи и отследить контаминацию до трех партий контаминированного MPA.
Одним из наиболее заметных примеров использования WGS для идентификации грибов и эпидемиологического отслеживания является появление грибков с множественной лекарственной резистентностью - Candida auris. Сообщалось, что C. auris вызывает смертельные инфекции и вспышки в больницах и учреждениях длительного ухода по всему миру. Известно, что C. auris трудно идентифицировать стандартными лабораторными методами из-за его сходства по внешнему виду и биохимическим характеристикам с другими кандидами. Обычные биохимические тесты обычно ошибочно идентифицируют C. auris как Candida haemulonii, а также с другими грибками, такими как Candida parapsilosus и Rhodatorula spp.. Существует обогащенный бульон, который можно использовать для преодоления ошибочной идентификации C. auris; однако для этого требуется 21 день инкубации с последующим получением агаровой культуры. Результаты WGS были использованы для подтверждения идентичности возбудителя и эпидемиологического мониторинга по всему миру.
Прогнозирование резистентности к противогрибковым препаратам с помощью WGS изучается у грибков, таких как Candida, но до недавнего времени не было опубликовано исследований по прогнозированию резистентности к противогрибковым препаратам грибков с помощью WGS. Тем не менее, многочисленные изоляты аскомицетного грибка Aspergillus fumigatus были исследованы с помощью WGS для изучения доминирующего механизма резистентности при мутациях гена, кодирующего белок, на который направлено действие триазольных противогрибковых препаратов.
WGS было проведено для 24 изолятов, как резистентных к азолам, так и чувствительных к ним, полученных из клинических и экологических источников из нескольких стран. Биоинформационный анализ SNPs высокого разрешения подтвердил мутацию TR34/L98H как единственный механизм резистентности к азолам. Используя этот подход, расширенные исследования 218 изолятов A. fumigatus, как клинических, так и из окружающей среды, из Великобритании и Ирландии, были изучены методом WGS для определения молекулярной эпидемиологии грибка и выяснения, происходит ли приобретение лекарственно-устойчивых изолятов группами риска.
Анализ данных подтвердил, что изоляты, резистентные к азолам, передаются из окружающей среды. Эти данные были использованы для проведения геномных исследований ассоциаций (GWAS - genome-wide association studies) и пангеномного анализа с целью выявления вариаций, связанных с резистентностью к итраконазолу, что позволило обнаружить потенциально новые механизмы резистентности, имеющие полигенную основу. Приведенные выше примеры иллюстрируют возможности WGS в изучении Aspergillus и резистентности к противогрибковым препаратам, однако существует множество примеров скоплений клинически значимых грибов в условиях клиники, где эта технология потенциально может раскрыть важные эпидемиологические проблемы инфицирования и распространения, а также резистентности к противогрибковым препаратам. Необходимо провести работу по изучению и внедрению в лабораториях соответствующих методов идентификации грибов с помощью WGS.
Полезной будет не только идентификация и эпидемиологическая информация, но и прогнозирование резистентности к противогрибковым препаратам с помощью WGS - это логичный следующий шаг, который может реально улучшить лечение грибковых инфекций.
Когда речь заходит о внедрении NGS-технологий в лаборатории клинической микробиологии, непосредственным применением NGS-технологии является WGS. WGS колоний непосредственно из культуры позволяет получить обширные знания, сопоставимые с обычным микробиологическим рабочим процессом. Однако WGS обеспечивает не только стандартную идентификацию и прогнозирование резистентности к противомикробным препаратам, но и возможность сравнения изолятов между собой для выявления вспышек заболеваний. Мы считаем, что внедрение WGS - это возможный первый шаг к внедрению NGS в лабораторию, поскольку он дополняет, а в некоторых случаях и улучшает обычный рабочий процесс микробиологической лаборатории.
4. Метагеномное секвенирование непосредственно образца
Если говорить о будущем NGS в клинической микробиологии, то главным преимуществом секвенирования метагеномов непосредственно из клинического образца является полное исключение процесса культивирования. Это может значительно сократить время выполнения исследований и предоставить медицинским работникам более своевременные ответы.
Существует два различных NGS-подхода к секвенированию метагеномов, которые могут быть использованы для обнаружения патогенов непосредственно из клинического образца (рис. 1). Первый - это процесс обогащения, известный как глубокое ампликонное или целевое секвенирование, при котором к выделенной ДНК применяются специфические праймеры для амплификации нужной группы патогенов (например, бактерий или грибов) или одного конкретного патогена, представляющего интерес (например, ВИЧ, SARS-CoV-2).
Второй подход известен как секвенирование методом дробовика (shotgun metagenomics), когда секвенируется вся выделенная ДНК или РНК из клинического образца, а работа по определению потенциального патогена выполняется в биоинформационном конвейере. Такой подход позволяет расширить сеть для поиска потенциальных патогенов, представляющих интерес.
В настоящее время еще нет одобренных Управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA) NGS-тестов для любого из этих подходов. Подобно WGS-тестам в лабораториях общественного здравоохранения, существуют лаборатории, сертифицированные в соответствии с Поправками к закону о совершенствовании клинических лабораторий (CLIA), в которых разработаны лабораторные тесты для прямого метагеномного секвенирования образцов. Существует несколько коммерческих вариантов метода дробовика, которые будут рассмотрены ниже.
4.1. Целевое (таргетное) секвенирование
Целевое секвенирование - это метод секвенирования, при котором процесс отбора или обогащения проводится для интересующего организма или группы организмов либо до, либо после процесса подготовки библиотеки (рис. 1). Существует несколько методов, которые могут быть использованы для выделения конкретного организма или группы организмов, представляющих интерес, и они включают в себя: ПЦР-амплификация и гибридизация зондов. Преимущество всех этих методов заключается в меньшем вмешательстве человеческой ДНК и более высокой чувствительности обнаружения в образцах с большим количеством человеческих клеток (например, в тканях или мокроте). Основным недостатком этих методов направленного секвенирования является ограниченное число патогенов, которые можно обнаружить. Целевое секвенирование непосредственно из образцов проводилось с использованием технологических платформ второго и третьего поколения.
ПЦР-амплификацию при целевом секвенировании также называют глубоким ампликонным секвенированием. Глубокое ампликонное секвенирование - это расширение технологии ПЦР, обеспечивающее более глубокий охват интересующего гена (генов). Наиболее известными применениями глубокого ампликонного секвенирования являются амплификация гена 16S рибосомальной РНК (16S рРНК) для идентификации бактерий и 28S рРНК или рибосомальных генов ITS для идентификации грибов.
Многие лаборатории утвердили и внедрили тесты для глубокого ампликонного секвенирования с идентификацией как бактерий, так и грибов, а одна группа даже продемонстрировала полезную схему, включающую оба метода. Кроме того, глубокое ампликонное секвенирование 16S облегчило идентификацию более трудно выращиваемых организмов, включая клещевые бактерии, которые обычно не обнаруживаются в обычных бактериальных культурах (например, Borrelia, Anaplasmsa, Ehrlichia и Rickettsia).
В частности, амплификация гена 16S была продемонстрирована в клинических условиях для различных типов образцов, включая суставную жидкость, кровь и спинномозговую жидкость (ЦСЖ). В случае перипротезных инфекций суставов (ППИС) обычная чувствительность бактериальной культуры, как известно, несовершенна из-за того, что пациенты получали предыдущие курсы антимикробной терапии. Было проведено сравнительное исследование пациентов с подозрением на ППИС для сравнения чувствительности целевого 16S-секвенирования с обычным бактериальным посевом, чтобы определить, улучшилась ли идентификация микроорганизма, вызвавшего инфекцию.
В общей сложности 47 ППИС локтевого сустава были положительными при секвенировании, и четыре из них (8%) были отрицательными при культивировании и положительными при целевом 16S-секвенировании. Всего было восемь случаев расхождения результатов между традиционной бактериальной культурой и целевым 16S-секвенированием, и они были получены в образцах ППИС, которые представляли собой полимикробные инфекции. В четырех случаях целевое 16S-секвенирование выявило дополнительные патогены. Эти данные отражают ценность и сложность целевого 16S-секвенирования, способного предоставить дополнительную информацию о таких ППИС, особенно в случаях, когда они являются культурально-негативными. Это исследование было проведено ретроспективно, но можно представить, что дополнительная информация, полученная в результате целевого 16S-секвенирования, могла бы изменить результаты терапии и состояние пациента.
Прямое глубокое ампликонное секвенирование образцов для идентификации грибов было изучено в ограниченной степени на свежих образцах и показало определенный успех. Однако единственный тип образцов, где глубокая ампликонная амплификация грибов показала свою перспективность и большую клиническую пользу, - это ткани с парафиновыми вкраплениями, фиксированные в формалине (FFPE).
Одним из традиционных методов выявления инвазивных грибковых инфекций является микроскопическая визуализация грибковых элементов в тканях FFPE в сочетании с положительным результатом культивирования. Однако нередки случаи, когда грибковые элементы обнаруживаются в тканях FFPE, но результат культивирования отрицательный или культура не была поставлена, поскольку это могла быть случайная находка. Кроме того, грибковые элементы, обнаруженные в тканях FFPE, могут быть неправильно идентифицированы в 21% случаев. Глубокое ампликонное секвенирование грибков в тканях FFPE может предложить врачам точную идентификацию увиденных организмов и позволить им применить правильный вариант противогрибковой терапии. В частности, было показано, что глубокое ампликонное секвенирование ITS позволяет идентифицировать грибковые элементы, обнаруженные в тканях FFPE, даже если на слайде присутствовало менее 50 грибковых организмов.
Метод глубокого ампликонного секвенирования используется в клинической вирусологической диагностике, в частности для определения резистентности к противовирусным препаратам у цитомегаловируса (ЦМВ) и вируса иммунодефицита человека (ВИЧ). Оба вируса имеют хорошо известные мутации в своих геномах, которые придают устойчивость к специфическим для каждого вируса противовирусным препаратам и подробно рассмотрены в других источниках. Коммерческое выявление известных маркеров резистентности к противовирусным препаратам с помощью секвенирования для ЦМВ - это тест, выполняемый как компанией Viracor, так и ARUP Laboratories. Лишь компания Viracor предлагает тест для определения профиля устойчивости к противовирусным препаратам с помощью секвенирования для ВИЧ-1 в дополнение к генотипированию интегразы ВИЧ-1.
Популярным подходом к целевому секвенированию, который используется при секвенировании генома человека, является обогащение зондов. В этом методе используются небольшие гибридизационные зонды, которые собираются в панели, включающие от 50 до миллионов зондов. В контексте секвенирования генома человека и панелей мутаций эти гибридизационные зонды добавляются к фрагментированной ДНК и обогащаются для выявления интересующих генов или мутаций.
Эта технология была усовершенствована для применения в клинической лаборатории с созданием панели гибридизации бактериальных зондов, известной как система секвенирования с захватом бактерий (BacCapSeq), и панели гибридизации вирусных зондов, известной как платформа секвенирования с захватом вирусов позвоночных (Virome Capture Sequencing Platform for Vertebrate Viruses (VirCapSeq-VERT). В настоящее время нет опубликованных работ, свидетельствующих о том, что эти специфические панели гибридизации зондов используются в клинической микробиологической диагностике.
Метод гибридизации зондов использовался в здравоохранении для обнаружения и отслеживания важных глобальных вирусных патогенов. В частности, в случае с вирусом Зика этот метод помог выявить линию и провести эпидемиологическое отслеживание вируса во время вспышки в середине 2010 года. Успех с вирусом Зика помог перенести этот подход на текущую пандемию SARS-CoV-2. Многие лаборатории общественного здравоохранения внедрили метод обогащения зондов для секвенирования SARS-CoV-2 непосредственно из представленных образцов, чтобы определить вариант, циркулирующий в обществе. Пока нет единого мнения о том, существует ли клиническая необходимость в генотипировании SARS-CoV-2 на уровне вариантов, но метод обогащения зондов делает это более достижимой возможностью, если лаборатория располагает соответствующими возможностями.
4.2. Секвенирование метагеномов методом дробовика
В отличие от целевого секвенирования, секвенирование метагеномов методом дробовика позволяет охватить более широкую сеть, поскольку секвенируются все нуклеиновые кислоты в образце (рис. 1).
Секвенирование всех нуклеиновых кислот позволяет выявить почти все патогены, включая бактерии, грибы, вирусы и паразитов, с помощью одного теста.
Этот метод секвенирования был успешно применен для выявления инфекции в различных образцах, включая обычно стерильные источники, такие как ЦСЖ, кровь и суставная жидкость. Кроме того, было продемонстрировано, что он помогает обнаружить инфекционный агент в образцах, в которых присутствует собственный микробиом, например, в образцах дыхательных путей, желудочно-кишечного тракта и мочи. Одним из ограничений метагеномного секвенирования методом дробовика является фон или интерференция нуклеиновых кислот человека или микробиома резидента, что может быть особенно опасно в таких образцах, как ткани или дыхательные секреты.
Этот подход был реализован в нескольких лабораториях в виде валидированного теста, и эти лаборатории служат клиническими референс-центрами для пациентов. Первая из них находится в сертифицированной по CLIA лаборатории клинической микробиологии Калифорнийского университета в Сан-Франциско и предлагает тест на метагеномное секвенирование ЦСЖ методом дробовика. Общая точность этого теста составляет 90%, а клиническая чувствительность и специфичность - 73% и 99% соответственно.
Имеются сообщения о различных случаях, свидетельствующих о диагностической пользе этого теста в случаях, когда все другие традиционные диагностические тесты были отрицательными, включая случай нейроцистицеркоза, вызванного инфицированием Taenia solium, случай вируса Западного Нила и случай нейробруцеллеза. С помощью этого теста было проведено годичное проспективное многоцентровое клиническое исследование пациентов с клинической картиной энцефалита, менингита и миелита, целью которого было определить полезность теста для диагностики этих заболеваний. С помощью метода дробовика в ЦСЖ было выявлено больше патогенов, чем при традиционном прямом выявлении в ЦСЖ (культура, анализ на антигены или экспрессные молекулярные методы).
Однако авторы не утверждают, что этот тест может заменить традиционные методы диагностики, но улучшить диагностический подход. Этот же метод метагеномного секвенирования был применен к другим жидкостям организма, включая абсцессы, суставную жидкость, перитонеальную жидкость, плевральную жидкость, бронхоальвеолярный лаваж (БАЛ) и мочу. Следует отметить, что в клинических лабораториях данного референс-центра нет возможности использовать данный метод секвенирования метагеномов методом дробовика для других биологических жидкостей. Единственный предлагаемый дробовой метагеномный тест - это исследование ЦСЖ.
Существует несколько коммерческих компаний, которые взяли за основу методологию дробовика и создали коммерческие анализы для различных источников организма. Первый из них, о котором мы расскажем, - это тест Karius для крови/плазмы. Этот тест, называемый тестом на свободную от клеток ДНК микроорганизмов или неинвазивной жидкой биопсией, был впервые представлен на рынке диагностики инфекционных заболеваний в 2016 году. Он предназначен для неинвазивного выявления глубоко залегающих инфекций и инфекций кровотока. В одном образце крови можно обнаружить >1250 мишеней различных бактериальных, грибковых, ДНК-вирусов и эукариотических паразитов.
В отчете для пациентов количество обнаруженных целей выражается в молекулах ДНК на микролитр (MPM). Результаты по обнаруженным мишеням представлены в графическом виде, что позволяет сравнить количество случаев обнаружения данной мишени программой Karius. В различных исследованиях было обнаружено и зарегистрировано более одной цели. Частота обнаружения нескольких патогенов различна, что подтверждается опытом авторов. Все мишени, обнаруженные в образце крови, представлены в иерархическом порядке, причем мишень с наибольшим содержанием, MPM, указана первой. Необходимо оценить результаты и использовать клиническое суждение, чтобы определить клиническую значимость некоторых обнаруженных мишеней. Показатели позитивности (%) для обнаруженных патогенов в представленных образцах варьируются в зависимости от возраста пациентов и от учреждения, но в среднем могут составлять 50-70%. При повышении дискриминации при заказе теста показатели позитивности могут быть более высокими.
Среди исследований, оценивающих совпадение положительных результатов секвенирования по Karius с обычными культурами, есть исследование, в котором изучались 2000 образцов, протестированных Karius, и было отмечено 97% совпадение положительных результатов секвенирования с результатами посева крови у пациентов с сепсисом. Существует еще несколько различных исследований, одни проспективные, другие ретроспективные, с относительно небольшим количеством пациентов, в которых совпадение результатов тестирования Karius и культуры или рибосомного секвенирования варьировало от 58 до 100%.
Приведенные ниже два случая демонстрируют широту применения теста для некультивируемых и культивируемых микроорганизмов и показывают полезность теста Karius при клинических инфекционных заболеваниях. Некоторые конкретные данные и описание были изменены для обеспечения анонимности пациентов.
Описание случая №1.
У маленького ребенка с синдромом Ди Джорджа через 2 недели после хирургического лечения порока сердца, все еще находившегося на аппарате ИВЛ, развились лихорадка и двусторонние легочные помутнения. Посевы крови и мокроты не выявили бактерий и грибов. Образец крови был отправлен на анализ по Karius, и через 48 часов был получен результат: 1365 MPM ДНК Pneumocystis jirovecii.
Пациенту была начата специфическая терапия против Pneumocystis jirovecii, состояние улучшилось, и он был отключен от аппарата ИВЛ. Через несколько дней анализ ПЦР на P. jirovecii в эндотрахеальном образце также оказался положительным. Последующий тест Karius через 2 недели был положительным на P. jirovecii в 65 MPM ДНК.
Описание случая №2.
Ребенок поступил с недельной повышенной температурой >38 С. При осмотре был обнаружен ранее не выявленный шум в сердце - аортальная регургитация, а эхокардиограмма показала двустворчатый аортальный клапан с большими вегетациями и разрывом клапана. Первоначально рутинные культуры крови были отрицательными. Через 36 часов тест Karius в плазме крови показал наличие Kingella kingae с 1890 МРМ ДНК. Были начаты антибиотики, и пациенту была проведена экстренная замена аортального клапана. Повторный тест Karius через 16 дней дал положительный результат на Kingella kingae с 55 MPM ДНК. Аэробные культуры крови оказались положительными через 9 дней инкубации, и тот же микроорганизм был выделен и идентифицирован с помощью масс-спектрометрии MALDI-TOF и секвенирования 16S рРНК.
Более поздние публикации о тесте Karius включают ретроспективный обзор использования теста в крупной детской больнице в Техасе, США. Авторы пришли к выводу, что в целом обычное микробиологическое исследование дает те же результаты, что и тест Karius, но с более коротким временем выполнения. Кроме того, у большинства пациентов при выявлении дополнительных микробных агентов с помощью теста Karius терапевтическое лечение не менялось. Напротив, проспективное исследование этого теста в сравнении с посевом крови и другими стандартными микробиологическими исследованиями у взрослых пациентов с лейкемией и фебрильной нейтропенией показало, что результаты теста Karius могли бы позволить более раннюю оптимизацию антимикробных препаратов у 47% пациентов.
Это было сравнительно небольшое исследование, включавшее 55 пациентов, но при оценке клинических данных было установлено, что причиной лихорадки у 87% пациентов была инфекция. По мнению многих экспертов в области инфекционных заболеваний, ценность теста Karius заключается в обнаружении неожиданных, трудно выявляемых или трудно культивируемых патогенов. Примером может служить обнаружение Rhizopus у одного пациента в вышеупомянутом исследовании в двух разных временных точках, когда на культивирование и идентификацию грибов уходило несколько дней.
Хотя повторное тестирование по Karius может быть проведено у пациента, оно обычно не повторяется, и нет контролируемых исследований, демонстрирующих преимущества серийного тестирования. В некоторых ситуациях, например при наличии трудно поддающегося лечению микроорганизма, вызывающего эндокардит или другие необычные инфекции, можно привести аргументы в пользу повторения теста, чтобы увидеть значительное снижение MPM после двух- или нескольких недель антимикробной терапии. Поскольку тест Karius очень дорог (2000 долларов за тест с дополнительными расходами, иногда значительными, в зависимости от учреждения), в некоторых медицинских центрах существуют процессы управления (например, консультации/проверка местными руководителями лабораторий в области диагностической микробиологии и инфекционных заболеваний взрослых и детей) для оценки необходимости проведения теста или любых последующих тестов Karius. Остается много нерешенных вопросов, касающихся использования и времени выполнения этого теста, значимости полимикробных результатов и того, насколько клинические результаты компенсируют высокую стоимость теста.
Еще одним коммерчески разработанным метагеномным тестом по методу дробовика является Explify™ Respiratory, который выполняется преимущественно на БАЛах и был выпущен компаниями IDbyDNA и ARUP Laboratories в 2017 году. Выделение РНК и ДНК из образцов БАЛ сопровождается подготовкой библиотек комплементарной ДНК и NG-секвенирования ДНК и секвенированием на приборах NextSeq или NovaSeq (Illumina) с медианной глубиной секвенирования 3-5 миллионов чтений. Explify™ Respiratory позволяет обнаружить >900 бактериальных, грибковых и вирусных респираторных патогенов. Обнаружение микроорганизмов основано на порогах обнаружения, установленных в ходе исследований по контролю качества, и результаты представляются полуколичественно. Результаты также стратифицируются и сообщаются как потенциальные патогены или дополнительные микроорганизмы на основе известной патогенности.
Этот клинический метагеномный NGS-анализ был применен для изучения пневмонии у различных групп пациентов, особенно у детей и взрослых с ослабленным иммунитетом. У таких пациентов часто наблюдаются загадочные пневмонические процессы, на которые не всегда дают ответ обычные бактериальные и грибковые культуры, а также однократные или мультиплексные ПЦР-анализы. Примером его полезности является выявление пропущенных патогенов у иммунокомпрометированных детей с пневмонией. Ранее пропущенные предполагаемые микробные патогены были выявлены в 18 из 41 (44%) БАЛ этих детей с угрожающей жизни пневмонией, включая 7 из 11 (64%) детей с фатальными инфекциями. Анализ Explify™ Respiratory выявил один патоген у 12 детей (63%), два - у 5 (26%) и четыре патогена - у 1 (5%) пациента. У этого количества детей были обнаружены бактериальные (13), грибковые (7) и вирусные (3) патогены.
Аналогичным образом, 30 иммунокомпрометированных взрослых с 31 эпизодом пневмонии прошли бронхоскопию и исследовали БАЛ с помощью теста Explify™ Respiratory, а результаты сравнивали с результатами традиционного микробиологического исследования (ТМИ). Окончательный микробиологический диагноз был поставлен в 11 случаях (35%) при использовании только ТМИ и в 18 случаях (58%) при использовании ТМИ и Explify™ Respiratory. Окончательный диагноз был поставлен в 20/31 случае (65%) только с помощью ТМИ и в 23/31 случае (74%) на основании ТМИ плюс Explify™ Respiratory, что не является большой разницей. Однако диагностическое преимущество анализа Explify™ Respiratory в основном заключалось в выявлении дополнительных бактериальных причин, а для диагностики грибковой пневмонии он оказался менее полезным в данном исследовании. Очевидно, что при интерпретации результатов Explify™ Respiratory, а также их значимости в контексте клинического состояния пациента необходимо руководствоваться клиническими суждениями. Иногда результаты анализа Explify™ Respiratory оказываются впечатляющими, но неожиданными; например, Pneumocystis jiroveccii (дрожжеподобный грибок) у пациента с ослабленным иммунитетом.
Примером потенциальной диагностической и клинической ценности теста Explify™ могут служить два взрослых пациента с ослабленным иммунитетом, страдающих пневмонией и отрицательными результатами обычных микробиологических исследований, у которых методом клинической метагеномики в БАЛ были обнаружены только Pantoea agglomerans или Ewingella americana. Рентгенологические данные соответствовали пневмонической патологии. Хотя это относительно редкие организмы, они могли способствовать развитию патологии, описанной у пациентов с ослабленным иммунитетом. Для диагностики загадочных пневмонических процессов, особенно у пациентов с ослабленным иммунитетом, целесообразно сочетать стандартные микробиологические диагностические подходы с данным анализом, чтобы дополнить клинические решения.
Наконец, коммерческая компания IDbyDNA расширила метагеномный подход, используемый в Explify™ Respiratory, на диагностику инфекций мочевыводящих путей с маркерами резистентности к противомикробным препаратам, и несколько соответствующих докладов были представлены на 32-м Европейском конгрессе по клинической микробиологии и инфекционным болезням, Лиссабон, Португалия, 23-26 апреля 2022 года. Эта панель идентификации патогенов в моче/антимикробной резистентности позволяет выявить >190 патогенов, включая 135 бактерий, 35 вирусов, 14 грибков и 7 паразитов, а также >2000 маркеров антимикробной резистентности. Ожидается, что дальнейшие подробности из рецензируемых работ расширят наше представление о применении и ценности этого метагеномного подхода для обнаружения патогенов в моче.
Применение метагеномики методом дробовика прошло долгий путь при различных инфекциях, описанных выше (менингит/энцефалит, сепсис, пневмония и инфекции мочевыводящих путей). Исследования, использующие этот подход для диагностики инфекций костей, связанных с ними тканей, а также собственных или протезированных суставов, ограничены. Данные для оценки относительной клинической ценности, влияния на клинический исход, чувствительности и специфичности недостаточны.
5. Факторы, которые следует учитывать при внедрении этих технологий
Что касается внедрения в лаборатории, то в настоящее время не существует утвержденных FDA США тестов для WGS, но многие лаборатории, сертифицированные CLIA, включая лаборатории общественного здравоохранения, имеют валидированные тесты для проведения WGS, включая и биоинформационные рабочие процессы. При принятии решения о том, является ли внедрение этих технологий секвенирования оптимальным для вашей лаборатории, необходимо учитывать множество факторов. Существует несколько отличных обзоров, в которых это обсуждается более подробно, но мы остановимся на нескольких основных факторах, которые мы считаем наиболее важными перед внедрением технологий секвенирования в клиническую микробиологию.
Первый фактор, который необходимо учитывать, - это процесс валидации лаборатории для этих NGS-приложений (т. е. WGS, целевой метагеномики и метагеномики дробовиком). Процесс валидации лабораторных тестов очень дорог и занимает много времени, что ограничивает круг лабораторий, которые могут использовать этот тип технологии. Параметры, необходимые для валидации лабораторных тестов, могут быть труднодостижимыми для таких приложений NGS.
Основные аспекты контроля качества лабораторных тестов для NGS трудно определить, поскольку в сообществе клинических микробиологов нет согласованного референсного стандарта. В случае с WGS легко определить последовательность штамма положительного типа и отрицательного контроля при каждом анализе, чтобы убедиться, что анализ работает правильно. Однако при использовании метагеномных подходов лабораториям придется собирать макет сообщества или пробы с интересующими организмами, и опять же нет согласия по поводу состава этих макетов сообществ.
Также трудно найти метод золотого стандарта для точного сравнения с результатами применения NGS, поскольку во многих случаях было показано, что применение NGS превосходит стандартные методы в обнаружении микроорганизмов. Недавно CDC США и Ассоциация лабораторий общественного здравоохранения США (APHL) создали веб-справочник, содержащий инструменты и ресурсы для систем управления качеством, к которым лаборатории могут обратиться при рассмотрении вопроса о валидации этих тестов (https://www.cdc.gov/labquality/qms-tools-and-resources.html). Однако даже при наличии этих инструментов и ресурсов сложность этих NGS-подходов и строгий процесс валидации в настоящее время удерживают эту технологию в пространстве референс-лабораторий и крупных академических медицинских центров.
Второй фактор, который необходимо учитывать при внедрении этих NGS-технологий, - это представление результатов, особенно в части интерпретации и клинической полезности метагеномных подходов.
Создание биоинформационных конвейеров для обработки данных о последовательностях требует значительного времени и опыта, причем не только для кодирования и вычислений, но и для микробиологической и клинической интерпретации результатов.
В настоящее время не существует коммерциализированных биоинформационных программ для интерпретации данных, однако несколько крупных академических медицинских центров опубликовали свои рабочие процессы для всех трех приложений NGS - WGS, целевого секвенирования и метагеномики методом дробовика. Один из аспектов, который мы поддерживаем и который несколько лабораторий рекомендовали для помощи в интерпретации и представлении результатов этих NGS-приложений, - это создание и использование группы лиц или консультативного совета. Члены группы/совета могли бы быть экспертами, помогающим в интерпретации интересных или неоднозначных результатов, и гарантировать, что сообщаемые результаты соответствуют клинической картине.
Последний важный фактор, который необходимо учитывать при внедрении этих NGS-подходов в лаборатории клинической микробиологии, - это стоимость. Хотя стоимость секвенирования образца или организма снизилась, затраты на внедрение этой технологии в лаборатории клинической микробиологии очень велики. Одним из первых препятствий, связанных с затратами, является наличие физического пространства для проведения лабораторной части анализа. Важно, чтобы лаборатория была приспособлена для раздельного хранения материалов и помещений до и после амплификации. Это поможет уменьшить возможную контаминацию образцов. Еще одной крупной статьей расходов, о которой уже упоминалось, является валидация тестов. Кроме того, после того как тест будет утвержден и внедрен, его трудно будет включить в лабораторию, чтобы заменить традиционные микробиологические культуры, которые обычно стоят не так дорого.
6. Будущее NGS в клинической микробиологии
Использование NGS в лаборатории клинической микробиологии уже стало реальностью, но только для немногих лабораторий, имеющих бюджет и персонал, позволяющий это сделать. В этом обзоре мы выдвигаем гипотезы и прогнозируем некоторые из потенциальных будущих направлений развития этой технологии в диагностической микробиологии. Во-первых, процесс секвенирования значительно подешевел с момента появления первого поколения секвенаторов. Можно предположить, что эта технология станет дешевле, что позволит ей быть более доступной для лабораторий, не входящих в состав крупных референс-лабораторий или крупных академических медицинских центров. Мы считаем, что секвенирование может стать недорогим и достаточно быстрым по сравнению с культурально-зависимыми методами. Например, с помощью WGS можно будет идентифицировать колонию, растущую на чашке, и получить АST-профиль этого организма за время, сравнимое с тем, которое требуется для проведения и простой идентификации при обычном микробиологическом исследовании.
Если рассматривать АST-тестирование, то основным критерием внедрения молекулярного AMR-тестирования в общее микробиологическое обследование является соответствие между фенотипическими и генотипическими результатами AMR.
Даже если в генотипическом профиле обнаружен маркер гена резистентности к антибиотикам, нет гарантии, что этот ген транслируется и транскрибируется для производства фермента или белка, обеспечивающего резистентность, если это не видно в результатах фенотипической чувствительности.
В будущем было бы полезно включить в программу исследования рабочий протокол протеомики, чтобы увидеть, какие белки экспрессируются, что поможет сделать вывод о фенотипических профилях чувствительности к антибиотикам.
Еще одна область, в которой в ближайшие 5 лет может быть достигнут значительный прогресс, - это биоинформационный анализ, в частности, для секвенирования метагеномов с методом дробовика. Существующие в настоящее время биоинформационные конвейеры для секвенирования методом дробовика позволяют обнаружить множество патогенов, присутствующих в данном образце. Однако современные технологии не позволяют собрать полный геном патогена, если только это не вирус. В будущем можно представить, что биоинформационный конвейер для проведения метагеномики методом дробовика в клиническом образце будет достаточно сложным, чтобы определить истинную сборку полного генома патогена с возможностью прогнозирования маркеров вирулентности и резистентности к противомикробным препаратам.
7. Заключение
Технологии NGS существуют уже несколько десятилетий, но только начинают менять диагностический потенциал лабораторий клинической микробиологии и здравоохранения. В этих областях WGS позволяет идентифицировать организмы и осуществлять надзор за потенциальными вспышками заболеваний. Кроме того, WGS помогает обнаружить не только известные гены и механизмы антимикробной резистентности, но и новые гены или механизмы, которые на данный момент не определены. Целевое и метагеномное секвенирование непосредственно из образцов помогает увеличить количество обнаруженных организмов, представляющих интерес, особенно тех, для которых обычные методы недостаточно чувствительны или недоступны. Эти технологии NGS меняют облик клинической микробиологической диагностики, какой мы ее знаем. Хотя NGS не может заменить обычные лабораторные исследования в области микробиологии, объем информации, получаемой с помощью секвенирования, только улучшит качество лечения пациентов.









